Comparative Gene Expression Analysis of Mouse and Human Cardiac Maturation
2016-11-17HidekiUosakiTaguchi
Hideki Uosaki*,Y-h Taguchi
1Division of Cardiology,The Johns Hopkins University School of Medicine,Baltimore,MD 21205,USA
2Department of Physics,Chuo University,Tokyo 112-8551,Japan
ORIGINAL RESEARCH
Comparative Gene Expression Analysis of Mouse and Human Cardiac Maturation
Hideki Uosaki1,*,a,Y-h Taguchi2,b
1Division of Cardiology,The Johns Hopkins University School of Medicine,Baltimore,MD 21205,USA
2Department of Physics,Chuo University,Tokyo 112-8551,Japan
Received 21 March 2016;revised 7 April 2016;accepted 10 April 2016 Available online 16 July 2016
Handled by Andreas Keller
Cardiac maturation;
Comparative gene expression analysis;
Microarray meta analysis;Principal component analysis;
Feature selection
Understanding how human cardiomyocytes mature is crucialto realizing stem cell-based heart regeneration,modeling adult heart diseases,and facilitating drug discovery.However,it is not feasible to analyze human samples for maturation due to inaccessibility to samples while cardiomyocytes mature during fetal development and childhood,as well as difficulty in avoiding variations among individuals.Using modelanimals such as mice can be a usefulstrategy;nonetheless,itis not well-understood whether and to what degree gene expression profiles during maturation are shared between humans and mice.Therefore,we performed a comparative gene expression analysis of mice and human samples.First,we examined two distinct mice microarray platforms for shared gene expression profiles,aiming to increase reliability of the analysis.We identified a set of genes displaying progressive changes during maturation based on principal component analysis.Second,we demonstrated that the genes identified had a differential expression pattern between adult and earlier stages(e.g.,fetus)common in mice and humans.Our findings provide a foundation for further genetic studies of cardiomyocyte maturation.
Introduction
Pluripotent stem cells(PSCs)hold tremendous potential for regenerative medicine,disease modeling,and drug discovery in a broad spectrum of tissue and cell types,such as cardiomyocytes[1-4].Recent advances in the field have rendered efficient and robust differentiation of cardiomyocytes from most of PSC lines[5-7].Although the maturation of differentiated cardiomyocytes into the adult-like stage is essential to study adult-onset diseases in vitro,fully matured cardiomyocytes have never been obtained[8].Moreover,there are no clear-cut and definitive markers available to evaluate cardiomyocyte maturation[8].Therefore,a detailed understanding of the cardiac maturation process in vivo is a prerequisite for further development of methods to maturate PSC-derived cardiomyocytes in vitro.
Uosaki et al.examined the detailed process of mice cardiac maturation using meta-microarray analysis[9].This and other studies demonstrated that the maturation of cardiomyocytes isa continuous process occurring during embryonic and postnatal development[9-12].Because of limited human samples obtained during the early life(potentially collected from aborted fetus,babies that died from accidents or other medical reasons,and/or biopsies from transplanted hearts)and technical difficulty in repetitive sample collection from the same individual,it is difficult to dissect the progression in humans from individual variations,e.g.,by measuring gene expression. Therefore,studies of cardiac maturation rely heavily on model animals,e.g.,mice.Here,the key question remain to be addressed is whether and to what extent cardiac maturation progresses are similar in mice and humans.
Comparative gene expression analysis[13]is a useful strategy to evaluate consistency between species.It enables studying multiple human diseases in mice,which are hard to investigate directly in humans[14].It can even help us to understand gene regulatory mechanisms in mammals using gene expression data from non-mammalian animals[15]. Moreover,it also helps in identifying highly-correlative expression profiles between putative orthologs across species[16].
In this study,we demonstrated the correlation of gene expression involved in cardiac maturation between mice and humans.We performed a meta-microarray analysis of data generated from mice samples ranging from the embryonic to the adult stages using two microarray platforms(Affymetrix Mouse Genome 430 2.0 Array,referred to as‘mouse 430 2.0”hereafter and Mouse Gene 1.0 ST Array,referred as‘mogene 1.0”hereafter)to collect a reliable set of genes correlating with the progression of cardiac maturation in mice.Subsequently,we evaluated whether highly-correlative expression profiles that were identified in the mice gene set exist in human samples.
Results
Performance comparison between frozen robust microarray analysis and microarray suite 5 method
In our previous paper[9],we employed the frozen robust microarray analysis(fRMA)[17]to analyze the gene expression profiles of more than 200 microarray datasets ranging from early embryonic to adult hearts.fRMA serves as a reliable platform to perform meta-microarray analysis[17]. Nonetheless,fRMA can only be applied to popular microarray platforms,such as mouse 430 2.0 and mogene 1.0,due to its requirement of preprocessed dataset.In addition,there is uncertainty on whether fRMA correctly performs batch effect extraction,although this is one of the primary reasons why fRMA is introduced.On the other hand,microarray suite 5 method(MAS5)is a method used for single-microarray preprocessing[18].We hypothesized that MAS5 can replace fRMA for meta-microarray analysis.
To evaluate the performance of MAS5 for data preprocessing,we collected 646 microarray datasets(Table S1)and preprocessed them with MAS5 as well as fRMA.To allow comparison,MAS5-processed data was log2 transformed and scaled(mean=0;standard deviation=1).Signal intensities of all 45,101 probesets on mouse 430 2.0 platform were well correlated between MAS5 and fRMA(R=0.90;Pearson correlation)(Figure 1A).Although probes with medium signal intensities(6-12 in fRMA)showed better correlation,more variability was observed for probes with lower or higher signal intensities.To evaluate whether this variability would compromise the overall analysis,we conducted principal component analysis(PCA)for signal intensities of preprocessed data by fRMA(Figure 1B)and MAS5(Figure 1C).The scatter plots of the first and second principal component(PC1 and PC2)values were almost identical.In addition,variable loadings for PC1 were well correlated between data preprocessed by fRMA and MAS5(R=0.89;Pearson correlation)(Figure 1D).These results suggest that MAS5 can replace fRMA for meta-microarray analysis.Therefore,data preprocessed by MAS5 were used for downstream analyses.As pointed out previously[9],PC1 represents the maturation process and PC2 seems to separate batch effects in either preprocessing method.
As PCA indicated a gradualmaturation process in the heart[9],we next assessed how gene expression changes during the maturation process.To detect gross changes,we averaged the signal intensities of each probe at each developmental stage for ranking.Figure 1E depicts the distribution of the intensity ranks.As expected,the majority of probesets at the early embryonic and adult stages ranked either first or fifth,whereas more than one third of the probes at the late embryonic stage ranked third,suggesting that the expression of each gene changes gradually and unidirectionally.This finding is important when considering the limited datasets of human heart samples,which are mostly early-gestation fetal and adult samples,for comparative genomics.
Probe-gene conversion
To perform comparative gene expression analysis,it is necessary to convert probesets to genes.In mouse 430 2.0,there were more than 45,000 probesets for 20,736 genes.We used mouse 4302.db to annotate probesets to genes.As a result,11,076 genes were annotated to single probesets,whereas the remaining genes were annotated to at least two probesets(Figure 2A).Seita et al.reported that identifying probes with the most dynamic ranges can be a good way to select probes[19].However,such a method might be vulnerable to noise. Therefore,we decided to choose probes based on the interquartile ranges(IQRs)rather than the full dynamic ranges.For instance,myomesin 2(Myom2),encoding an M-protein that is expressed in mature cardiomyocytes[20],was annotated to 4 different probesets(Figure 2B).One probeset(1438372_at)showed a very small dynamic range,whereas the other three probesets displayed similar but distinct patterns,with the widest IQR observed for the 1457435_x_at probeset.Different from Myom2,Slc2a1that encodes glucose transporter 1(Glut1)was annotated to 3 probesets(Figure 2C),which share similar IQRs.In contrast to mouse 430 2.0,more than 95%(19,925 out of 20,915 in total)of genes were annotated to a single probeset in mogene 1.0 when using mogene10st trans criptcluster.db to annotate probesets to genes(Figure 2D).Therefore,for the mogene 1.0 data,we simply averaged the signal intensities from multiple probesets to obtain the expression level of a particular gene.
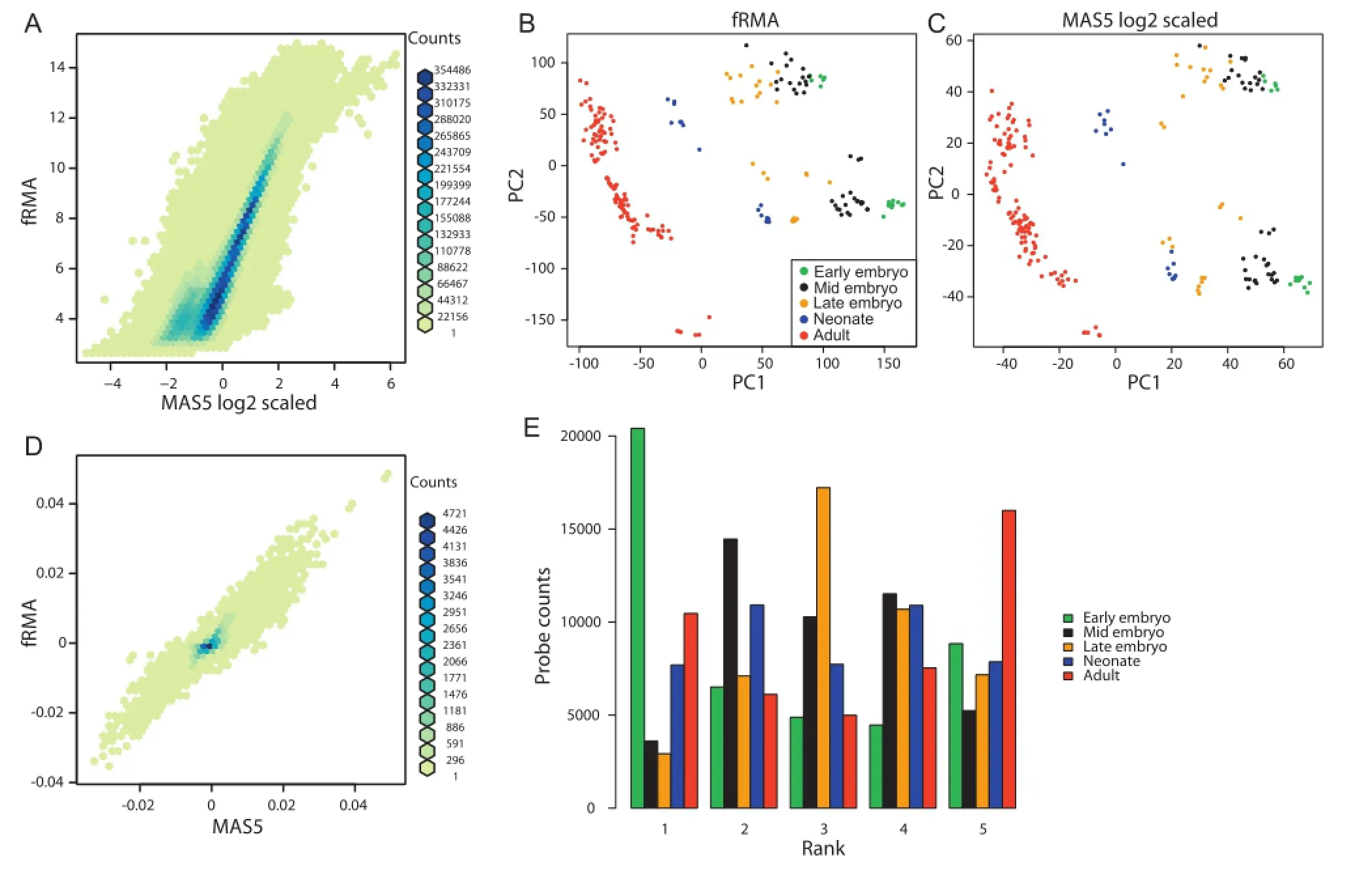
Figure1 Comparison of MAS5 and fRMA for mouse 430 2.0 array data preprocessing
Identification of mice genes associated with cardiac maturation using PCA
Next,we used PCA to identify genes associated with cardiac maturation in mice.As shown in Figure 1C with probe-level PCA,PCA clearly distinguished the samples from different stages(Figure 3A).Neonatal samples were grouped into two clusters.Notably,one neonatal cluster close to the late embryonic stage and the other cluster close to the adult stage included samples from postnatal day(P)3 and P7,respectively,supporting the notion that PC1 is an explanatory variable for cardiac maturation.Similarly,we also performed PCA for the mogene 1.0 data(Figure 3B).For some unknown reasons,data for some samples from a single institute were widely divergent from the other datasets.Therefore,these samples were excluded from entire analysis(data not shown,marked as‘GSI”in Table S2).Although the number of samples for each stage was small and plots were sparse,the overall patterns for PCA plots were similar between the mouse 430 2.0 array data and mogene 1.0 data.
To identify genes associated with cardiac maturation,we first plotted PC1 loadings of each gene for mouse 430 2.0 and mogene 1.0 data(Figure 3C).The loadings were well correlated(R=0.78).Next,we added the individual loadings for each gene.As the summed loadings followed a normaldistribution(data not shown),we selected genes with loadings higher than mean+2 standard deviation(SD)and lower than mean-2SD as genes that are significantly associated with cardiac maturation(colored in blue and red,respectively,in Figure 3C).As more than 3600 genes were unique to either array(Figure 3D),we also determined significant genes for each ofthe two arrays(Figure 3E and F).In total,we identified 648 genes,including 293 and 355 genes associated with mature and immature status,respectively(full lists available in Table S3).
Characterization of the maturation-associated genes
A linear model was employed to examine whether the genes identified above followed the trajectory of maturation(Figure 1E).First,we averaged the signal intensities of genes across samples of certain stages,which changed gradually with progressing stages for both mouse 430 2.0(Figure 4A)and mogene 1.0(Figure 4B).We next conducted the linear regression analysis for each gene to obtain P values and calculated false discovery rates(FDRs)in order to adjust for multiplecomparisons.Approximately 98%and 89%of the identified genes in the mouse 430 2.0 and mogene 1.0 arrays,respectively,had an FDR<0.10,suggesting linear gene expression alterations for most of the genes identified.
To further characterize biological properties of the identified genes,we performed KEGG pathway analysis with DAVID[21,22].Pathways with an FDR<0.01 were considered significant(nodes in color,Figure 4C and D,Table S4 and S5).For the genes associated with immature status,ribosome-and cell cycle-related(e.g.,DNA replication and oocyte meiosis)pathways were significantly enriched(mmu03010:ribosome;mmu04110:cell cycle,Figure 4C,Table S4).On the other hand,for the genes associated with mature status,oxidation and mitochondria-related pathways(mmu05012:Parkinson’s disease;mmu00190:oxidative phosphorylation;mmu05010:Alzheimer’s disease;mmu05016: Huntington’s disease;mmu00020:citrate cycle or TCA cycle)and cardiac pathways(mmu04260:cardiac muscle contraction;mmu05414:dilated cardiomyopathy,DCM,and mmu05410: hypertrophic cardiomyopathy(HCM))were significantly enriched(Figure 4D,Table S5).Taken together,these findings indicate that the genes identified are associated with cardiac maturation.
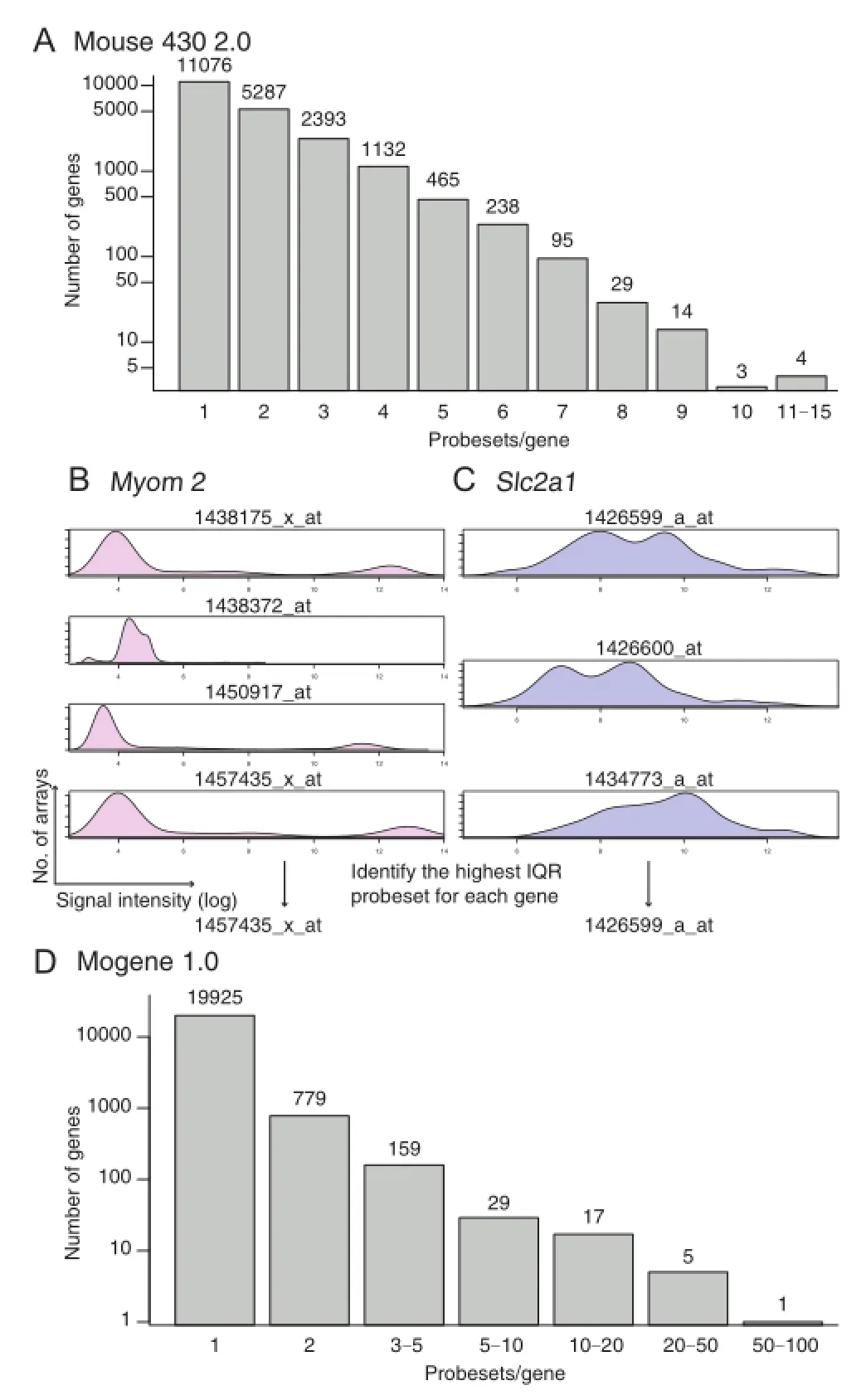
Figure2 Probeset-to-gene symbolconversion
Comparison with human datasets
Finally,we assessed the expression patterns of the genes identified in mice in human datasets.We found two distinct datasets of human hearts including fetal and adult hearts(GSE62913 and GSE71148)[22,23].GSE62913 contains RNA-seq data obtained from fetalventricles and atria,as well as adult hearts.We performed PCA with all genes as well as with the maturation-associated genes,respectively.Among the 648 maturation-associated mice genes identified above,we found human counterparts of 520 genes in the GSE62913 dataset(234 and 286 for mature and immature status,respectively).PCA with all genes as well as with maturation associated genes similarly revealed distinctive patterns between fetal samples and adult hearts(Figure 5A and B).The other dataset GSE71148 is an Illumina HumanHT-12 V4.0 expression beadchip dataset for fetal and adult heart samples.We identified 586 maturation-associated genes conserved between humans and mice(262 and 324 for mature and immature status,respectively).Consistent with the PCA on GSE62913,PCA on GSE71148 with all genes or the maturationassociated genes both generated patterns distinctive between fetal and adult samples(Figure 5C and D).
To assess whether gene expression patterns in mice and humans are correlated and whether the usage of maturation associated genes improves the correlation over the usage of all genes,we compared expression changes in mice and humans using all genes or the maturation-associated genes only(Figure 5E-H).As the human fetal heart samples were from fetus in the first and second trimesters(7-20 weeks),we used early embryonic mice hearts for comparison.We found that expression changes between adult and early embryo/fetus using all genes showed good correlation between mice and humans for mouse 430 2.0 dataset(R=0.49,Figure 5E)and mogene 1.0 dataset(R=0.51,Figure 5G).Nonetheless,the gene expression changes of maturation-related genes alone showed better correlation for both datasets(R=0.73 for mouse 430 2.0,Figure 5F and R=0.78 for mogene 1.0,Figure 5H).Overall,286 out of 324 immature status-associated genes and 237 out of 262 mature status-associated genes showed higher expression in fetal and adult hearts,respectively.Interestingly,most of the genes that showed inconsistency with the findings in mice did not show significant differences between fetal and adult heart samples in humans(only 8 genes showing more than 1.5-fold changes,Table S6).It is of note that MYH7 was among the immature-associated genes identified in the mice,and was highly expressed in human adult hearts as is widely known.

Figure3 Selection of genes associated with cardiac maturation
Taken together,gene expression pattern of cardiac maturation between early embryonic/fetal and adult stages is mostly consistent across species,and the maturation-related genes identified in mice can be mostly recapitulated in humans.
Discussion
In this study,we identified cardiac maturation-associated genes in mice based on PCA of data from two distinct mice microarrays.We demonstrated that the expression of the genes identified change progressively during maturation and that the expression patterns are well conserved between mice and humans.Although mice and human adult cardiomyocytes are different in terms of cell size,length of action potential,and beating rate,etc.,they share some common features e.g.,morphology,abundant mitochondria,and sarcomere structure[8].Our findings indicate that mice and humans follow a similar maturation process.MYH6 and MYH7,the genes encoding alpha and beta myosin heavy chains,are differentially expressed in mice and humans.Myh6 encodes a predominant form of myosin heavy chain in adult mice heart and Myh7 isexpressed in embryonic mice heart,whereas opposite expression pattern of these two genes is found in humans[24,25]. In accordance herewith,our comparative gene expression analysis successfully identified that MYH7 is a gene associated with immature stage in mice,but highly upregulated in human adult hearts.
Cells derived from either mice model or mice/human PSCs are often used for maturation studies.However,PSC-derived cardiomyocytes barely mature[9].More importantly,there are no established readouts to define maturation status of cardiomyocytes.Structural and functional readouts,which include cellsize,morphology,t-tubule formation,calcium handling,action potential,and mitochondrial function,are often used[26-28].It is known how morphology and structure change during maturation in mice or rat but it is unknown for human.Physiological features were only studied for adult cardiomyocytes but not for embryonic and neonatal cardiomyocytes.Therefore,these readouts cannot be used to measure maturation status quantitatively at this point.The gene list we provided(Table S3)could serve as a resource for developing defined,objective,and reliable readouts,as expression of these genes change monotonically during maturation in both mice and humans.
As we used PCA-based gene selection and made a comparison only between the adult and early embryonic/fetal stages,some of the highly differentially-expressed genes shown in Figure 5E and G were not selected based on PCA.Thus,we took an alternative approach for gene selection to evaluate whether the genes that are highly differentially expressed between adult and early embryo/fetus are sufficient to recapitulate the heart maturation pattern.Briefly,we summed the human and mice differential signal intensities of each gene.As the summed differential signal intensities followed a normal distribution,we selected genes for which expression levels fell out of the range of mean±2SD(Figure S1A and S1B).Although only one third of the alternatively selected genes overlapped with the genes selected using the PCA-based method(Figure S1C and S1D),the PCA patterns generated with the alternatively selected genes were similar to those generated with all genes(Figure S1E-H).As we demonstrated in Figure 1E as well as Figure 4A and B,the maturation process in the heart is unidirectional,and most genes related to maturation changed progressively.Therefore,the genes highly differentially expressed between the adult and early embryoic/fetal stages successfully represented the maturation process,which would be more appropriate for finding specifically-expressed genes.PCA granted unidirectional change and would be more appropriate for studying the process of maturation.
Finally,in this study,we also tackled a bioinformatics issue—the limitations of fRMA.Although fRMA was designed to avoid batch effects by using frozen data sets generated from a large quantity of datasets,fRMAdid not outperform MAS5,which is a single array-based normalization method.Our results demonstrate that the performance of fRMA is correlated well with that of MAS5,suggesting that MAS5 can be used in place for fRMA.

Figure4 Characterization of the genes associated with cardiac maturation
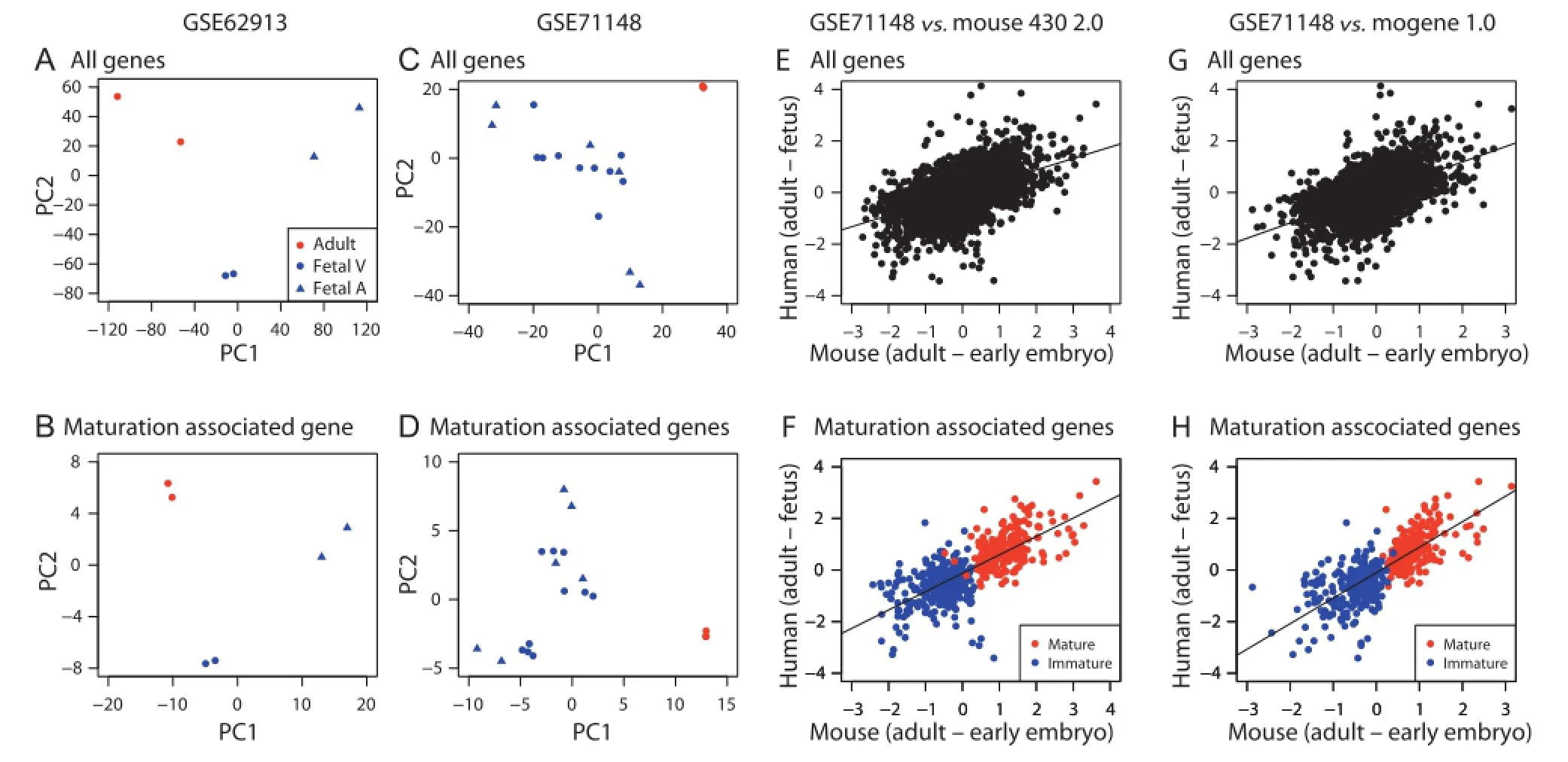
Figure5 Comparison of mice datasets with human datasets
Conclusions
In this study,we performed a comparative gene expression analysis of mice and human cardiac maturation.As a result,we identified more than 500 genes that share distinct expression patterns during cardiac maturation between mice and humans.These genes could be further explored for their potential as genetic markers to investigate cardiomyocyte maturation in future.
Methods
mRNA expression
All mRNA expression profiles analyzed in this study were downloaded from the Gene Expression Omnibus(GEO,http://www.ncbi.nlm.nih.gov/geo/).Mouse 430 2.0 gene expression profile was selected from profiles analyzed in our previous study[9].Detailed information about mouse 430 2.0 and mogene 1.0 arrays is listed in Tables S1 and S2,respectively.Profiles analyzed in Figures 1,3 and 4 were generated from five developmental stages with sample numbers(N)provided for mouse 430 2.0 and mogene 1.0,respectively.These include early embryonic(embryonic day(E)8-11,N=16 and 12),mid embryonic(E12-15,N=39 and 4),late embryonic(E16-18,N=26 and 2),neonate(postnatal day(P)1-10, N=16 and 2),and adult(>4-week old,N=115 and 134)stages.Only wild-type and non-treated samples were included in the current study.Human gene expression profiles were taken from GSE62913 and GSE71148.GSE62193 contains RNA-seq data for human PSC-derived cells,as well as fetal and adult hearts,whereas GSE71148 comes from an Illumina array transcriptome study for 20 samples from fetal and adult hearts,including Ref-pool(GSM1828516).
Preprocessing
Multiple preprocessing methods were employed in this study.‘MAS5-scale”indicates scaling was performed after MAS5 preprocessing,while‘MAS5-log2-scale”indicates that a log2 transformation was performed before scaling but after MAS5 preprocessing.
fRMA
fRMA was conducted using the Bioconductor/R fRMA package.Annotation packages mouse4302frmavecs and mogene.1.0.st.v1frmavecs were used for mouse 430 2.0 and mogene 1.0 arrays,respectively.
MAS5
MAS5 normalization was conducted for mouse 430 2.0 and mogene 1.0 data by using the MAS5 function in the Bioconductor/R affy and xps packages,respectively.
Scaling and log2 transformation
Scaling,which extract means and normalize standard deviation to one,was performed with the scale function in R.Additionally,log2 transformation was also performed using R.
To convert probesets to genes,we identified probesets with the highest IQRs of signal intensity for mouse 430 2.0.To determine the IQR,we analyzed 429 arrays for brain,212 arrays for heart,142 arrays for kidney,and 137 arrays for liver.All arrays were preprocessed with fRMA and the IQR was determined for each probeset.The probe-gene match list was used to convert MAS5-preprocessed data.The conversion table is available as Table S7.As only less than 5%of genes were annotated to multiple probesets in mogene 1.0(Figure 2D),we simply averaged the signal intensities of multiple probesets for a particular gene.
Human datasets
Read countdata of GSE62193 were scaled to normalize the individual samples(mean=1 and standard deviation=0),while normalized and log2-transformed data for GSE71148 was directly obtained from GEOand used for subsequentanalysis.
PCA
PCA was conducted using the prcomp function in Rto demonstrate overall differences of samples.
Identification of maturation-associated genes
Maturation-associated genes were identified using two different approaches.For genes common to the mouse 430 2.0 and mogene 1.0 arrays,PC1 loadings of each array were summed.Genes with summed PC1 loadings more than a mean+2SD or less than a mean-2SD were selected as maturation-associated genes.On the other hand,for genes unique to either of arrays,genes with PC1 loadings more than a mean+2SD or less than a mean-2SD of the corresponding array were selected.
Developmental stage wide coarse-grained gene expression analysis
孙静等[13]用半枫荷抗炎有效部位的全提取物(A组分)、齐墩果酸提取物纯化品(B组分)和除去齐墩果酸的提取物(C组分)处理以HBV-DNA转染的人肝细胞株HepG2,结果发现A、B组分对乙肝病毒的HBeAg与HBsAg抗原均具有很好的抑制作用,而C组分对抗原无抑制作用,因此判断对病毒抗原具有抑制活性的成分为齐墩果酸。
In this analysis,we employed MAS5 preprocessed profiles generated from the mouse 430 2.0 array.Average of expression of the i-thgene at each developmental stage,xis,was defined as xis≡where s is one of five aforementioned developmental stages and Nsis the number of samples that belong to the stage,xijis expression of the i-th gene in j-th samples.Averaged values were subsequently ranked across stages.
Linear regression analysis of developmental-stage coarse-grained gene expression
Regression analysis was done using the following equation: xis=ais+bi,where aiand biare the regression coefficients,and s takes values 1-5 corresponding to the developmental stages in the order of early,mid,late,neonatal,and adult,respectively.The linear regression analysis was carried out using lm function in R[29].P values were adjusted to meet FDR criterion using the fdrtool function in the fdrtool[30]package.Regressions with q values(adjusted P values)<0.1 were regarded to be significant.
KEGG pathway enrichment analysis
Enrichment analysis for KEGG pathways was performed by uploading gene symbols to DAVID.Numbers of genes overlapping between KEGG pathways were used as weights to generate KEGG path way net works shownin Figure 4C and D with the igraph[31]package in R[29].
Mapping of mice genes to human genes
Identical official gene symbols found in mice and human data were considered as a pair and used for comparison in Figure 5.
Authors’contributions
HU and YHT planed the research project;HU performed all analyses.Both HU and YHT were involved in manuscript writing,read and approved the final manuscript.
Competing interests
The authors have declared that there are no competing interests.
Acknowledgments
HU was supported by Maryland Stem Cell Research Fund,USA(Grant No.2015-MSCRFF-1765).YHT was supported by the grants from the Ministry of Education,Science(grant No.KAKENHI 26120528)and Chuo University joint research grant.
Supplementary material
Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j. gpb.2016.04.004.
[1]Inoue H,Nagata N,Kurokawa H,Yamanaka S.IPS cells:a game changer for future medicine.EMBO J 2014;33:409-17.
[2]Matsa E,Burridge PW,Wu JC.Human stem cells for modeling heart disease and for drug discovery.Sci Transl Med 2014;6:239ps6.
[3]Onder TT,Daley GQ.New lessons learned from disease modeling with induced pluripotent stem cells.Curr Opin Genet Dev 2012;22:500-8.
[4]Cho GS,Fernandez L,Kwon C.Regenerative medicine for the heart:perspectives on stem-cell therapy.Antioxid Redox Signal 2014;21:2018-31.
[5]Laflamme MA,Chen KY,Naumova AV,Muskheli V,Fugate JA,Dupras SK,et al.Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts.Nat Biotechnol 2007;25:1015-24.
[6]Uosaki H,Fukushima H,Takeuchi A,Matsuoka S,Nakatsuji N,Yamanaka S,et al.Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression.PLoS One 2011;6: e23657.
[7]Burridge PW,Matsa E,Shukla P,Lin ZC,Churko JM,Ebert AD,et al.Chemically defined generation of human cardiomyocytes. Nat Methods 2014;11:855-60.
[8]Yang X,Pabon L,Murry CE.Engineering adolescence:maturation of human pluripotent stem cell-derived cardiomyocytes.Circ Res 2014;114:511-23.
[9]Uosaki H,Cahan P,Lee DI,Wang S,Miyamoto M,Fernandez L,et al.Transcriptional landscape of cardiomyocyte maturation. Cell Rep 2015;13:1705-16.
[10]Di Maio A,Karko K,Snopko RM,Mejı´a-Alvarez R,Franzini-Armstrong C.T-tubule formation in cardiacmyocytes:two possible mechanisms?J Muscle Res Cell Motil 2007;28:231-41.
[11]Ziman AP,Go´mez-Viquez NL,Bloch RJ,Lederer WJ.Excitation-contraction coupling changes during postnatal cardiac development.J Mol Cell Cardiol 2010;48:379-86.
[12]Vreeker A,van Stuijvenberg L,Hund TJ,Mohler PJ,Nikkels PG,van Veen TA.Assembly of the cardiac intercalated disk during pre-and postnatal development of the human heart.PLoS One 2014;9:e94722.
[13]Kozian DH,Kirschbaum BJ.Comparative gene-expression analysis.Trends Biotechnol 1999;17:73-8.
[14]Tseveleki V,Rubio R,Vamvakas SS,White J,Taoufik E,Petit E,et al.Comparative gene expression analysis in mouse models for multiple sclerosis,Alzheimer’s disease and stroke for identifying commonly regulated and disease-specific gene changes.Genomics 2010;96:82-91.
[15]Kobayashi I,Ono H,Moritomo T,Kano K,Nakanishi T,Suda T.Comparative gene expression analysis of zebrafish and mammals identifies common regulators in hematopoietic stem cells. Blood 2010;115:e1-9.
[16]Mangelsen E,Kilian J,Berendzen KW,Kolukisaoglu UH,Harter K,Jansson C,et al.Phylogenetic and comparative gene expression analysis of barley(Hordeum vulgare)WRKY transcription factor family reveals putatively retained functions between monocots and dicots.BMC Genomics 2008;9:194.
[17]McCall MN,Bolstad BM,Irizarry RA.Frozen robust multiarray analysis(fRMA).Biostatistics 2010;11:242-53.
[18]Rajagopalan D.A comparison of statistical methods for analysis of high density oligonucleotide array data.Bioinformatics 2003;19:1469-76.
[19]Seita J,Sahoo D,Rossi DJ,Bhattacharya D,Serwold T,Inlay MA,et al.Gene expression commons:an open platform for absolute gene expression profiling.PLoS One 2012;7:e40321.
[20]Schoenauer R,Lange S,Hirschy A,Ehler E,Perriard JC,Agarkova I.Myomesin 3,a novel structural component of the M-band in striated muscle.J Mol Biol 2008;376:338-51.
[21]Huang DW,Sherman BT,Lempicki RA.Bioinformatics enrichment tools:paths toward the comprehensive functional analysis of large gene lists.Nucleic Acids Res 2009;37:1-13.
[22]Huang DW,Sherman BT,Lempicki RA.Systematic and integrative analysis of large gene lists using DAVIDbioinformatics resources.Nat Protoc 2009;4:44-57.
[23]van den Berg CW,Okawa S,Chuva de Sousa Lopes SM,van Iperen L,Passier R,Braam SR.Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 2015;142:3231-8.
[24]Lompre´AM,Nadal-Ginard B,Mahdavi V.Expression of the cardiac ventricular alpha-and beta-myosin heavy chain genes is developmentally and hormonally regulated.J Biol Chem 1984;259:6437-46.
[25]Everett AW.Isomyosin expression in human heart in early preand post-natal life.J Mol Cell Cardiol 1986;18:607-15.
[26]Kuppusamy KT,Jones DC,Sperber H,Madan A,Fischer KA,Rodriguez ML,et al.Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes.Proc Natl Acad Sci U S A 2015;112:E2785-94.
[27]Feaster TK,Cadar AG,Wang L,Williams CH,Chun YW,Hempel JE,et al.Matrigel mattress:a method for the generation of single contracting human-induced pluripotent stem cell-derived cardiomyocytes.Circ Res 2015;117:995-1000.
[28]Lee DS,Chen JH,Lundy DJ,Liu CH,Hwang SM,Pabon L,et al. Defined microRNAs induce aspects of maturation in mouse and human embryonic-stem-cell-derived cardiomyocytes.Cell Rep 2015;12:1960-7.
[29]R Core Team.R:a language and environment for statistical computing.R Foundation for Statistical Computing:Vienna,Austria.2015.
[30]Klaus B,Strimmer K.fdrtool:estimation of(local)false discovery rates and higher criticism.2015.R package version 1.2.15.
[31]Csardi G,Nepusz T.The igraph software package for complex network research.InterJournal 2006;Complex Systems:1695.
*Corresponding author.
E-mail:huosaki1@jhmi.edu(Uosaki H).
aORCID:0000-0002-8964-8609.
bORCID:0000-0003-0867-8986.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.04.004
1672-0229©2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
猜你喜欢
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Personalized Computer Simulation of Diastolic Function in Heart Failure
- Comparison of Cox Model Methods in A Low-dimensional Setting with Few Events
- Absent MicroRNAs in Different Tissues of Patients with Acquired Cardiomyopathy
- Profiling and Validation of the Circular RNA Repertoire in Adult Murine Hearts
- The Role of Quality Control in Targeted Next-generation Sequencing Library Preparation
- Long non-coding RNA Databases in Cardiovascular Research