RP-HPLC法测定防风通圣丸中栀子苷、芍药苷、黄芩苷、大黄素的含量*
2016-11-15杨小军方博文丁永辉
杨小军,方博文,丁永辉
1西北民族大学化工学院,甘肃 兰州 730124;2甘肃省食品药品监督管理局
RP-HPLC法测定防风通圣丸中栀子苷、芍药苷、黄芩苷、大黄素的含量*
杨小军1,方博文1,丁永辉2
1西北民族大学化工学院,甘肃 兰州 730124;2甘肃省食品药品监督管理局
目的:建立RP-H PLC同时测定防风通圣丸中栀子苷、芍药苷、黄芩苷和大黄素含量的方法。方法:采用高效液相色谱(RP-H PLC)梯度洗脱,在不同波长下测定含量。色谱柱:Zorbax Eclipse X D B-C18键合硅胶色谱柱(4.6 m m×150 m m,5 μm);柱温为室温;流动相为乙腈-2%磷酸水溶液,采用梯度洗脱;流速1.0m L/m i n;检测波长分别为240 nm(栀子苷、芍药苷),275 nm(黄芩苷、大黄素)。结果:栀子苷、芍药苷、黄芩苷和大黄素的进样量分别在0.056~0.56 g(r=0.999 7),0.103 5~1.029 7 g(r=0.999 2),0.207 4~3.128g(r=0.999 3),0.04~0.423g(r=0.999 6)范围内线性关系良好,平均加样回收率分别为99.14%,99.65%,99.03%,99.16%;RSD分别为1.08%,1.01%,0.85%,0.86%。结论:该方法简便、快速、灵敏、精密度高、重现性良好、结果准确,可用于同时测定防风通圣丸中栀子苷、芍药苷、黄芩苷和大黄素的含量。
RP-H PLC;防风通圣丸;栀子苷;芍药苷;黄芩苷;大黄素;含量测定
防风通圣丸(水丸)为收载于《中华人民共和国药典》的成方制剂[1],由防风、荆芥、薄荷、麻黄、大黄、芒硝、栀子、滑石、桔梗、石膏、川芎、当归、白芍、黄芩、连翘、甘草、白术(炒)17味中药材组成。其中芍药苷、黄芩苷、栀子苷及大黄素为处方中主要有效成分。有报道[2-5]采用高效液相色谱法对防风通圣丸(水丸)中黄芩苷和大黄素的含量测定,但同时测定其中栀子苷、芍药苷、黄芩苷和大黄素含量的方法未见文献报道,本研究采用双波长RP-H PLC法对处方中栀子苷、芍药苷、黄芩苷及大黄素的含量进行测定,为提升防风通圣丸质量标准及含量测定的方法提供科学依据。
1 材料与方法
1.1 试验仪器与试药 Agilent 1200高效液相色谱仪(美国Agilent公司);配置真空脱气机(G1322A);自动进样器(G1329A);真空二极管阵列检测器(DAD,G1315B);四元泵(G1311A),Zorbox Eclipse XDB-C18键合硅胶色谱柱(4.6m m×150m m,5 μm);Agilent 1200色谱工作站(美国Agilent公司);AB204-S型电子分析天平(瑞士Mettler Toledo公司);AP001-0671型抽滤机;AS3120型双频数控超声波清洗仪(天津奥特赛斯恩斯仪器有限公司);DFT-200型万能粉碎机(温岭市林大机械有限公司);甲醇(分析纯,天津市北方天医化学试剂厂);甲醇(色谱纯天津市光复精细化工研究所,公司);乙腈(色谱纯,天津市凯信化学工业有限公司);磷酸(分析纯,烟台市双双化工有限公司);水为超纯水。栀子苷对照品(中国药品生物制品检定所,批号:749-8801);芍药苷对照品(中国药品生物制品检定所,批号:110736-201333);黄芩苷对照品(中国药品生物制品检定所,批号:110715-201316);大黄素对照品(中国药品生物制品检定所,批号:110756-200110);防风通圣丸(水丸)(甘肃众友药业制药有限公司,批号:90903、90904、90905,规格6g×10丸)。
1.2 溶液的制备
1.2.1 对照品溶液的制备 取栀子苷对照品、芍药苷对照品、黄芩苷对照品和大黄素对照品,精密称定后,分别置于10 m L容量瓶中,加一定量甲醇(分析纯),摇匀,置超声仪中超声至全部溶解,放冷至室温,加甲醇至刻度,摇匀,即得含栀子苷、芍药苷、黄芩苷及大黄素对照品溶液。然后分别精确量取上述各对照品贮备液按体积1∶1混合,充分摇匀,即得含栀子苷56μg/m L,芍药苷100μg/m L,黄芩苷106 μg/m L,大黄素49 μg/m L的对照品混合溶液,取适量混合对照品溶液,用微孔滤膜(孔径∶0.45μm)滤过,弃去初滤液,取续滤液,备用。
1.2.2 样品溶液的制备 取防风通圣丸约3 g,剪成小块,精密称定,置研钵中,以40 m L甲醇分次研磨,定量转移至50 m L具塞容量瓶中,超声提取1小时,取出放冷至室温后加甲醇定容至刻度,摇匀,静置,取上清液,用微孔滤膜(孔径∶0.45 μm∶有机系)滤过,弃去初滤液,取续滤液,备用。
1.2.3 阴性对照品溶液的制备 栀子苷、芍药苷、黄芩苷及大黄素分别来源于处方中栀子、芍药、黄芩及大黄药材,按防风通圣丸(水丸)的处方中各药味的比例和制备工艺,分别制备缺少栀子、缺芍药、缺黄芩、缺大黄的阴性对照样品,按照“1.2.2项”下供试品溶液配制方法制备各阴性对照样品溶液,即得。
1.3 色谱条件 色谱柱:Zorbax Eclipse XDB-C18键合硅胶色谱柱(4.6 m m×150 m m,5 μm);柱温:室温;检测波长:240 nm(栀子苷、芍药苷)、275 nm(黄芩苷,大黄素);流动相:乙腈(A)-2%磷酸溶液(B),梯度洗脱程序见表1。
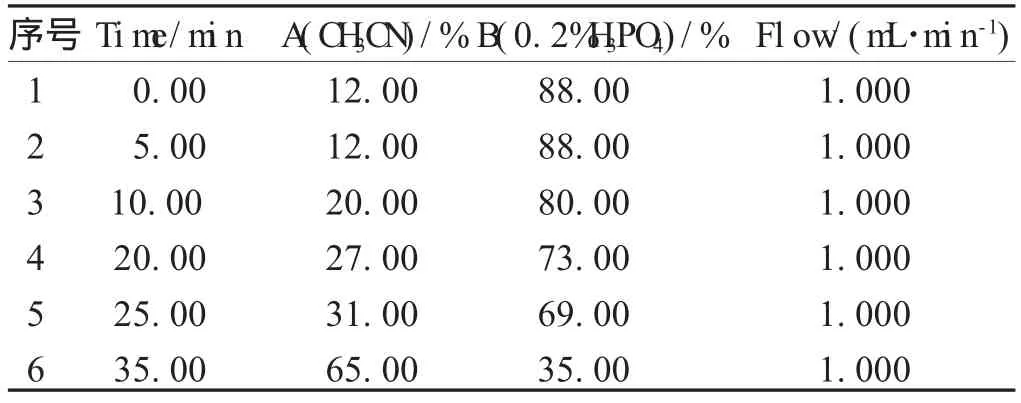
表1 梯度洗脱程序
1.3.1 分离度考察 分离度是用来判断物质在色谱柱中的分离情况,常用分离度作为色谱柱分离能力的指标,用R表示。其中R=2(tR1-tR2)/(W 1+W 2)。精密吸取上述防风通圣丸样品溶液,按上述色谱条件进样,进样量10 μL,结果见表2。
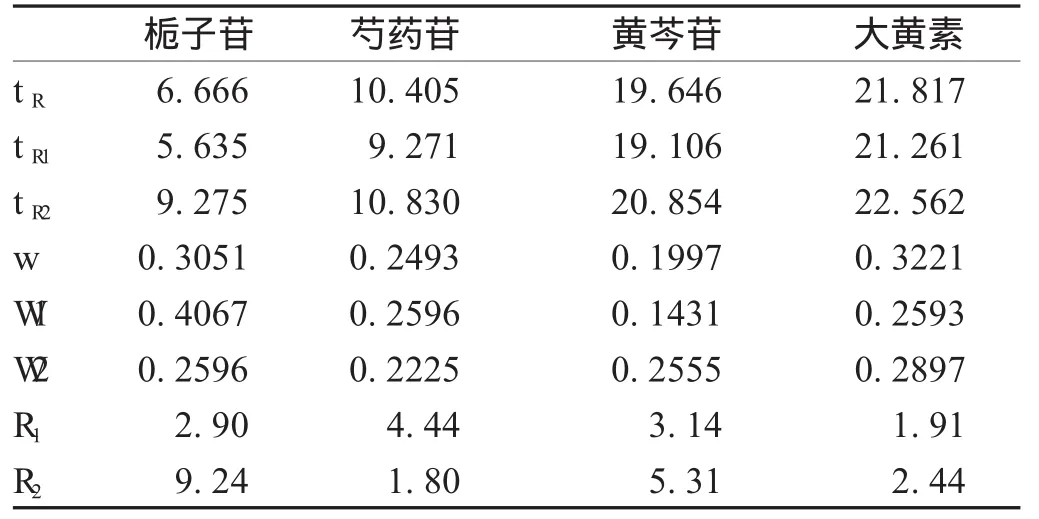
表2 分离度考察结果
由表2可见,栀子苷、芍药苷、黄芩苷和大黄素与相邻杂质峰之间的分离度均大于1.5,表明4种待测组分均能与杂质完全分离,分离效果良好。
1.3.2 专属性实验 精密吸取对照品混合溶液、供试品溶液、各阴性对照溶液,按上述色谱条件,分别进样分析,记录色谱图,结果在供试品溶液色谱图中,有与对照品混合溶液色谱图中栀子苷、芍药苷、黄芩苷及大黄素保留时间一致的特征峰,保留时间分别为6.71、10.428、19.712、33.512分钟。并且供试品峰之间基线较平稳分离度好,阴性对照样品色谱图中相应位置上无此特征峰,说明阴性对照样品对检测无干扰,结果表明本法的专属性良好,见图1—3。
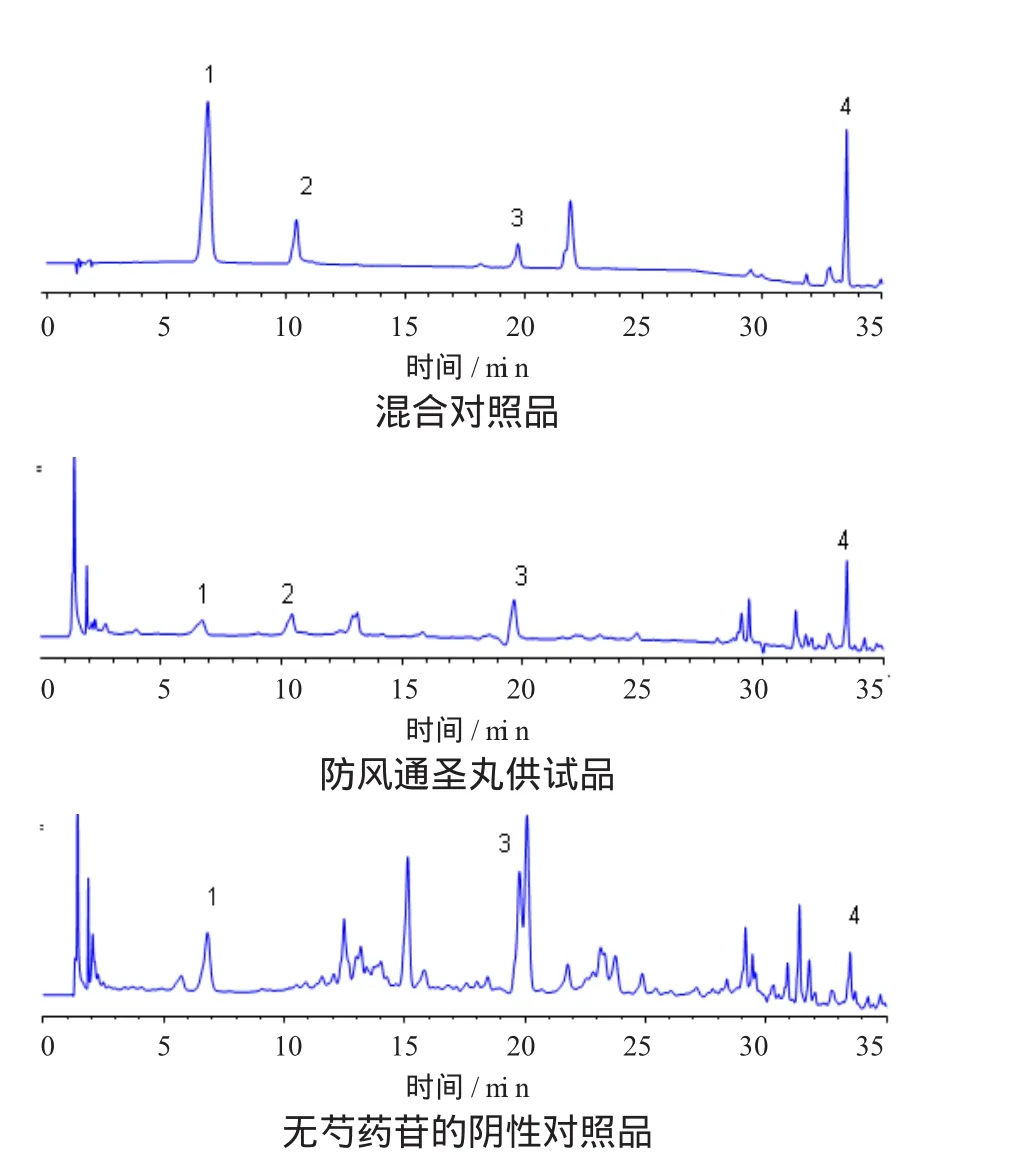
图1 在240 nm处的H PLC图谱
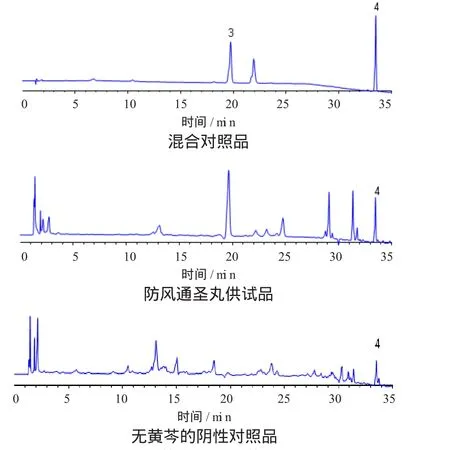
图2 在275 nm处的H PLC图谱
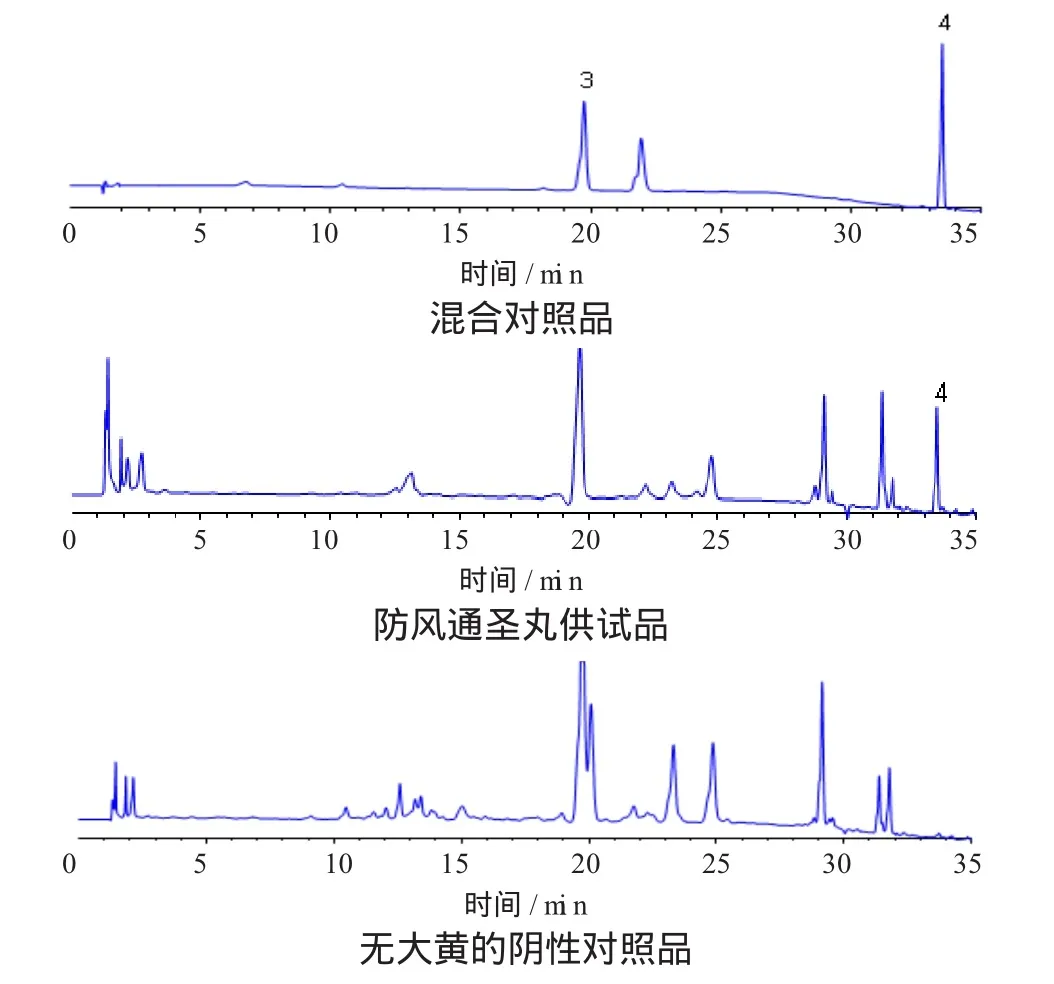
图3 在275 nm处的H PLC图谱
1.3.3 拖尾因子实验 检查待测峰的拖尾因子是为了保证色谱柱的分离效果和测定精度。拖尾因子是通过计算5%峰高处峰宽与峰顶点至前沿的距离来评价峰形的参数,用T表示。其计算公式为:T=W 0.05h/2d1。式中W 0.05h为5%峰高处的峰宽;d1为峰顶点至峰前沿之间的距离。
精密吸取样品溶液,按上述色谱条件进样,进样量10μL,结果见表3。
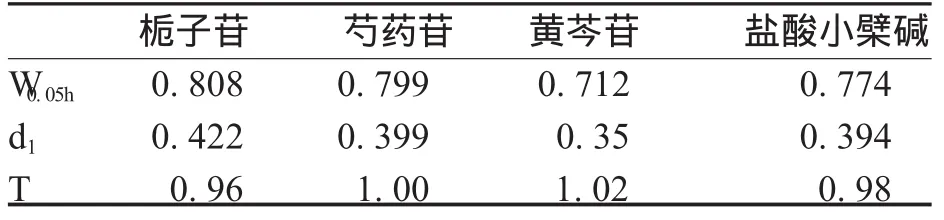
表3 拖尾因子计算结果
由表3可见,栀子苷、芍药苷、黄芩苷和大黄素的拖尾因子介于0.95~1.05之间,符合《中国药典》规定,峰形良好。
1.3.4 理论板数计算 理论板数N用于评价色谱柱的效能,是色谱的柱效能参数之一。N取决于固定相的性质(粒度、粒径分布等)、填充状况、色谱柱柱长、流动相的种类好流速及测定柱效所用物质的性质。若峰形对称,则N可用下式表示:

式中tR表示保留时间,W h/2表示半高峰宽。
精密吸取上述混合对照品溶液,按上述色谱条件进样,进样量为10μL,结果见表4。
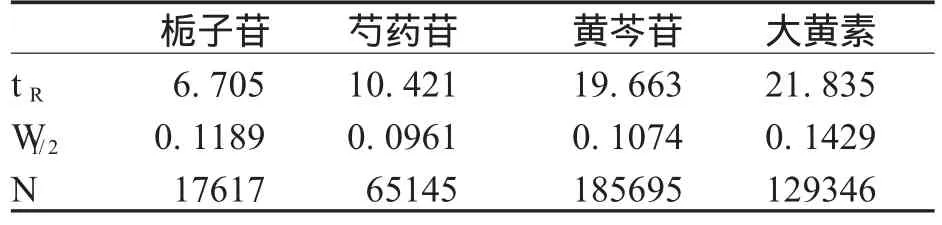
表4 理论板数计算结果
理论板数栀子苷为17 617,芍药苷为65 145,黄芩苷为185695,大黄素为129346。
2 结果
2.1 标准曲线与线性关系考察 精密吸取对照品混合溶液2,4,6,8,10,15,20 μL,按“1.3”项下色谱条件,进样测定,记录色谱图,以峰面积Y为纵坐标,以对照品进样量X(μg)为横坐标绘制标准曲线。栀子苷、芍药苷、黄芩苷及大黄素的标准曲线见图4—7。
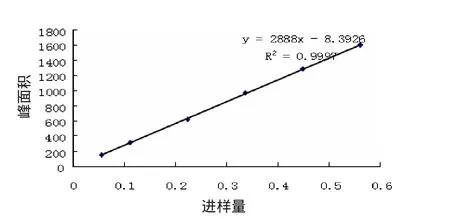
图4 栀子苷标准曲线
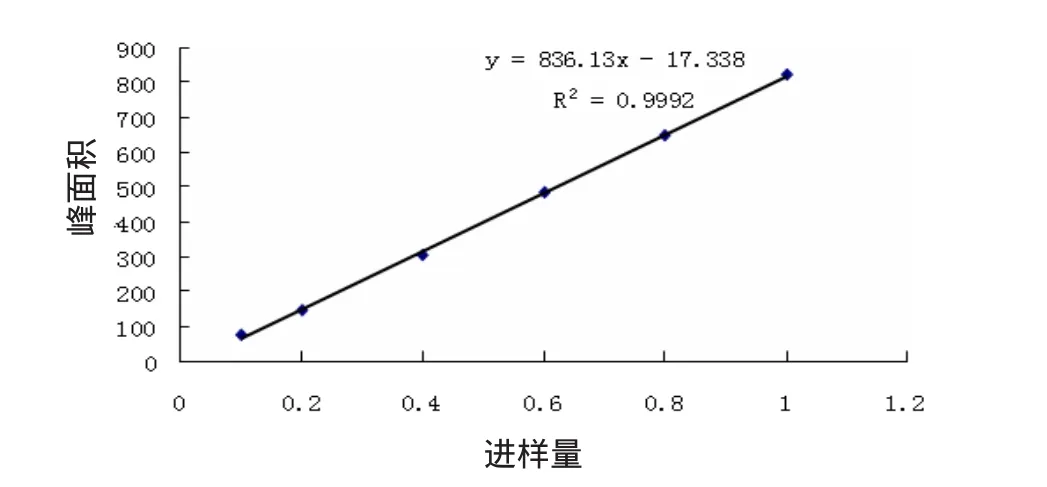
图5 芍药苷标准曲线
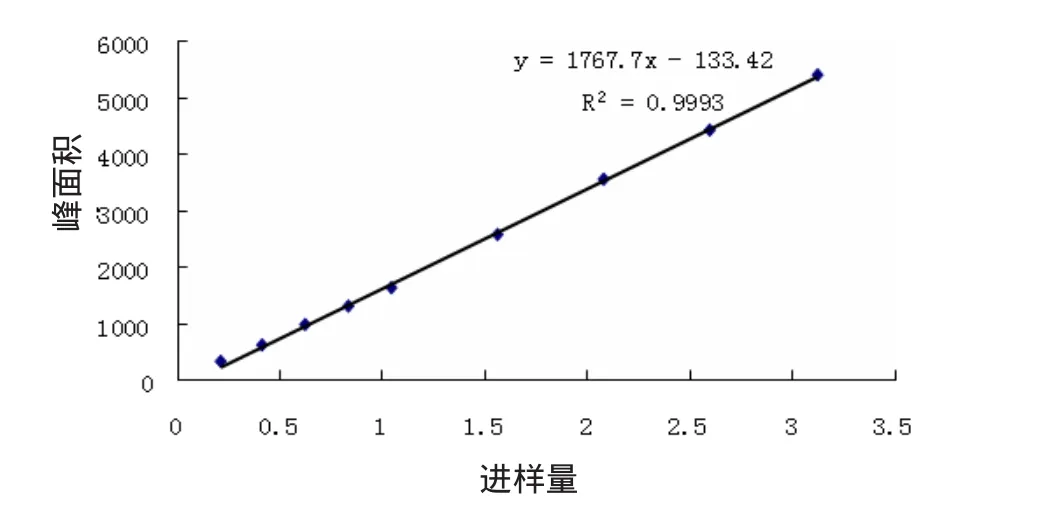
图6 黄芩苷标准曲线
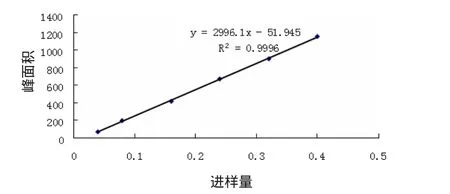
图7 大黄峰标准曲线
进行线性回归得回归方程:栀子苷:Y=2 888.0X-8.392 6,r=0.999 7,线性范围:0.056~0.56 μg;芍药苷:Y=836.13X-17.338,r=0.999 2,线性范围:0.103 5~1.029 7,黄芩苷:Y=1767.7X-133.42,r=0.999 3,线性范围:0.207 4~3.128 μg;大黄素:Y=299.1X-51.945 4,r=0.999 6,线性范围:0.04~0.423 μg。表明栀子苷,芍药苷、黄芩苷及大黄素的线性关系良好,见表5。
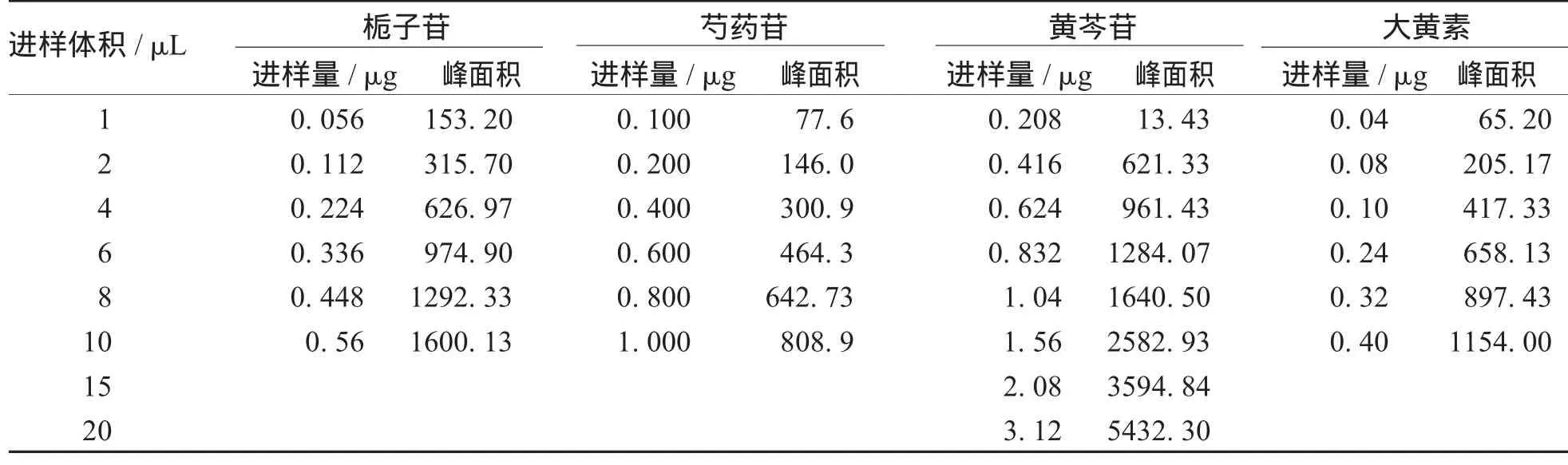
表5 栀子苷、芍药苷、黄芩苷及大黄素的标准曲线
2.2 精密度实验 精密吸取混合对照品溶液,按“1.3”项下色谱条件,连续进样6次,记录色谱图,进样量10 μL,栀子苷、芍药苷、黄芩、大黄素峰面积RSD(%)分别为1.23%、0.93%、1.02%和0.90%。表明仪器精密度良好,见表6。
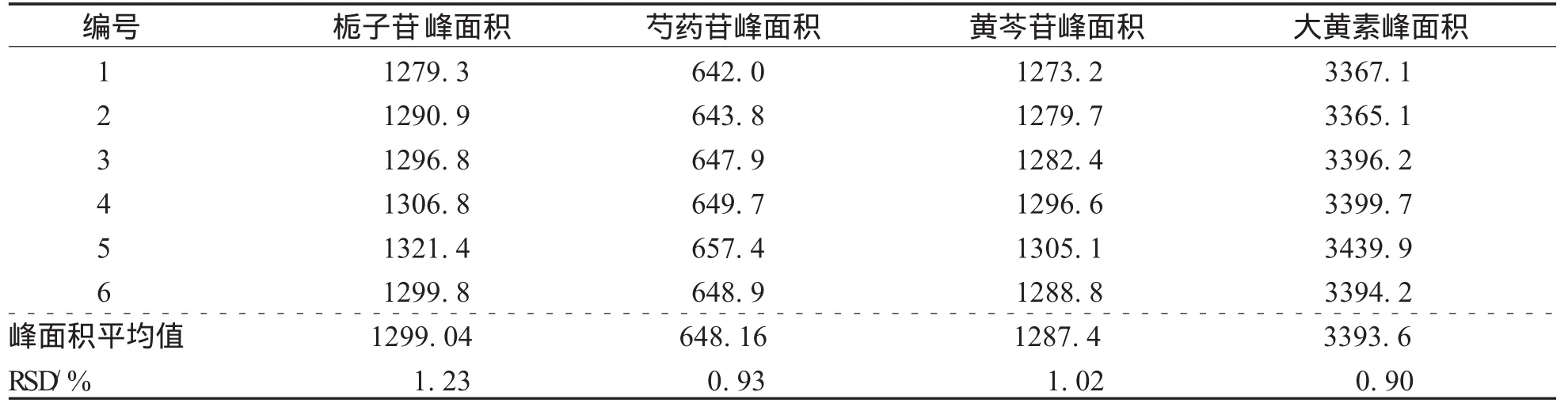
表6 精密度考察结果(n=6)
2.3 重复性实验 取同一批号的防风通圣丸供试品(批号:90903)6份,每份约3g,分别精密称定,按“1.2”项下的制备方法平行制备供试品溶液6份,按“1.3”项下的色谱条件分别进样,进样量为15μL,记录色谱峰面积,计算各成分含量。
栀子苷、芍药苷、黄芩苷及大黄素的平均含量分别为0.584 2,0.755 2,5.672 0,0.628 9 m g/g,RSD分别为1.76%,2.21%,0.58%及0.91%。表明方法的重现性良好,结果见表7。

表7 重复性考察结果(n=6)
2.4 稳定性实验 取配制好的同一供试品溶液(批号:90903),室温下放置,每2小时进样一次,进样量为15 μL,按“1.3”项下的色谱条件进行进样分析,并记录各时间下栀子苷、芍药苷、黄芩苷及大黄素的色谱峰面积,计算得峰面积RSD依次为1.81%、1.68%、0.58%、1.43%。表明本供试品在24小时内基本稳定,见表8。
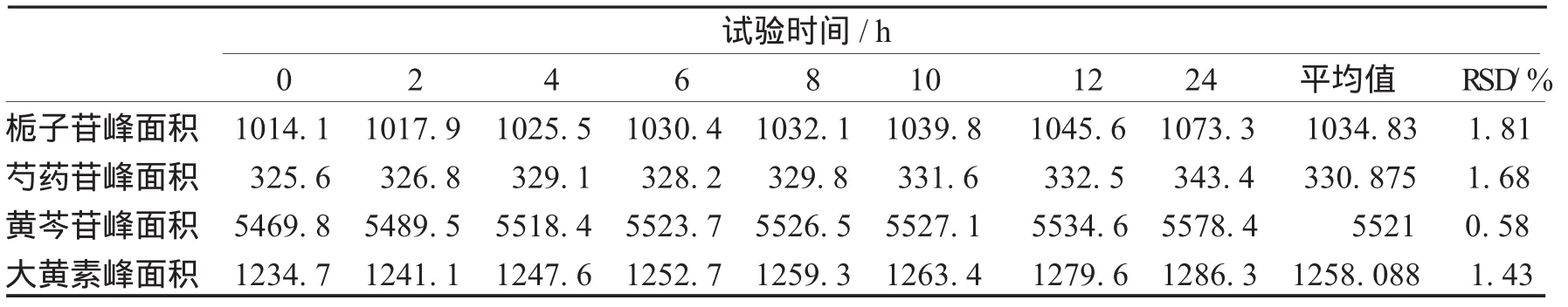
表8 稳定性考察结果
2.5 加样回收率实验 精密称取已知含量的同一批号(批号:90903)防风通圣丸(水丸)6份,每份约3.0 g,按“1.2”项下的制备方法制备供试品溶液,分别精密量取上述溶液1.0 m L,置于5 m L容量瓶中,分别加入栀子苷对照品溶液(含栀子苷56 μg/m L)0.15m L、芍药苷对照品溶液(芍药苷100 μg/m L)0.2 m L、黄芩苷对照品溶液(黄芩苷106 μg/m L)0.4 m L、大黄素对照品溶液(大黄素49 μg/m L)0.15 m L,混合均匀,按上述含量测定项下的方法进行测定,进样量为15 μL,记录各色谱峰面积。按下式进行回收率计算,结果见表9。
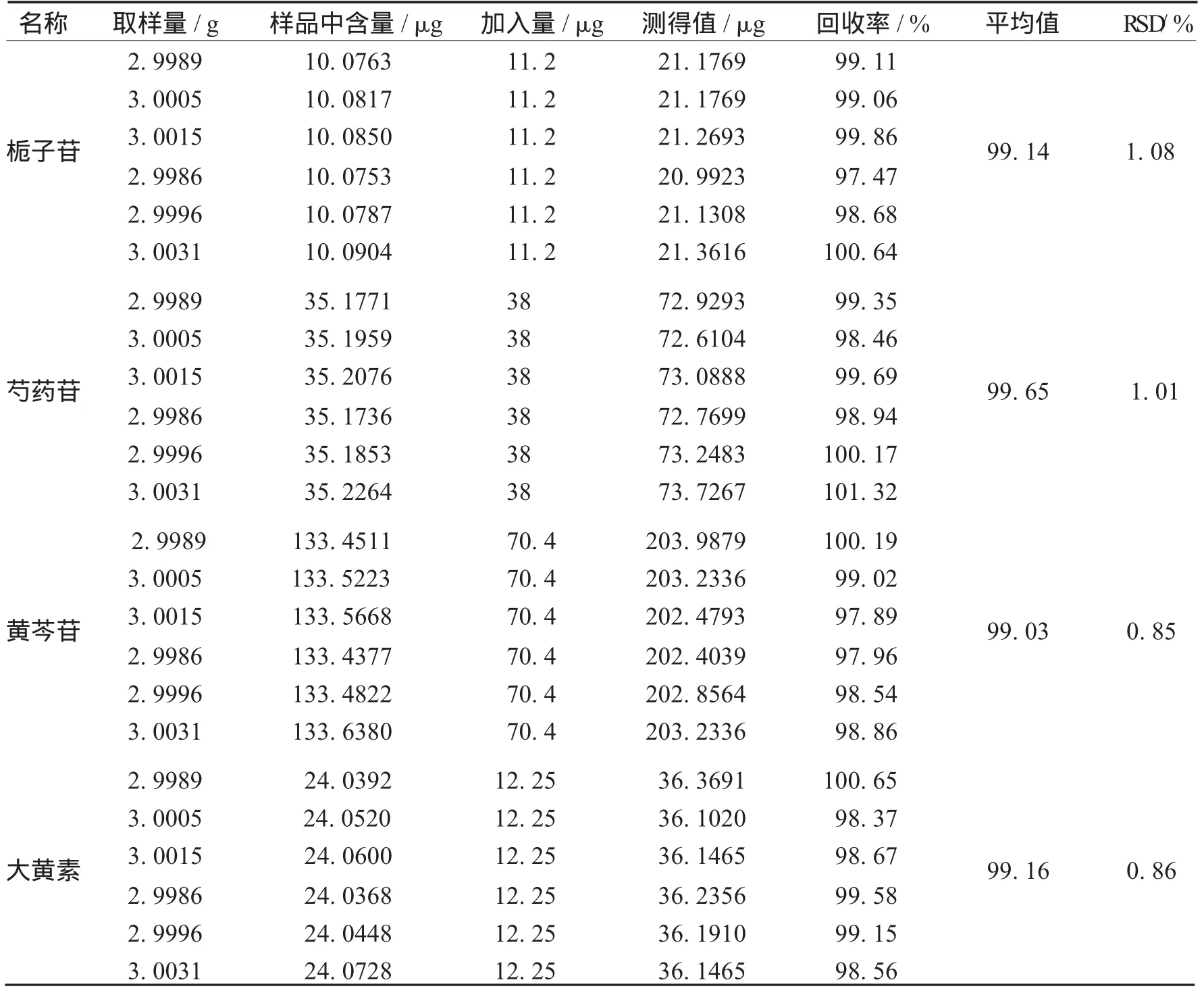
表9 加样回收率实验结果(n=6)
回收率(%)=(测得值-样品中含量)/加入量×100%
2.6 样品含量测定 分别取3个不同批号的防风通圣丸(水丸)各3份,每份约3 g,精密称定,按“1.2”项下样品的制备方法平行制备各样品溶液,在上述色谱条件下进样测定,进样量15 μL,在240 nm、275 nm、波长处分别记录栀子苷、芍药苷、黄芩苷及大黄素的色谱峰面积,根据线性回归方程积计算防风通圣丸中栀子苷、芍药苷、黄芩苷及大黄素的含量,结果见表10。

表10 防风通圣丸(大蜜丸)含量测定结果
其中以防风通圣丸(水丸)(批号:90903)计算,取样量为2.998 9 g的一组数据为例,分别计算栀子苷、芍药苷、黄芩苷和大黄素的相关数据。由于样品待测成分浓度在其线形范围之内,根据样品配置过程和栀子苷、芍药苷、黄芩苷和大黄素的线性回归方程计算:
已知:栀子苷、芍药苷、黄芩苷和大黄素的峰面积分别为:Y=428.7,423.5,3406,1028.7。
1)批号090903样品中15μL样品栀子苷含量为:
X=(Y+8.3926)/2888.0 =(483.7+8.3926)/2888.0=0.1704 μg
因此样品中所含栀子苷为:
m1=0.170 4×50/15/2.9989=0.1682m g/g 2)芍药苷的含量为:
X=(Y+17.338)/836.13
=(423.5+17.338)/831.43=0.5302μg
因此样品中所含芍药苷为:
m2=0.5302×50/15/2.9989=0.5865m g/g 3)黄芩苷的含量为:
X=(Y+133.43)/1767.7
=(3406+133.43)/1767.7=2.0023μg
因此样品中所含黄芩苷为:
m3=2.0023×50/15/2.9989=2.225m g/g
4)大黄素的含量为:
X=(Y+51.945)/2996.1
=(1028+51.945)/2996.1=0.3686 μg
因此样品中所含大黄素为:
m4=0.3686×50/15/2.9989=0.4008m g/g
其他组的数据算法同上,计算结果见表10。
3 讨论
3.1 流动相的选择 实验过程中发现,在不同时间段内,改变流动相中乙腈的含量,即随着乙腈含量的增加,选择性增强,分离效果较明显。但是流动相中乙腈的比例过高会导致基线的飘移,拖尾因子的效果较明显。由于样品中所测成分的极性不同,因此考虑运用梯度洗脱,在不同的时间段内,逐渐提高乙腈的比例,使各成分先后出峰,找到最佳的有效成分分离条件。本实验中最终选择了乙腈-2%磷酸水溶液作为流动相,采用梯度洗脱程序,可使栀子苷、芍药苷、黄芩苷和大黄素与其他成分离开,且分离度好,拖尾因子效果较差。
3.2 检测波长的选择 本实验中3种物质在240 nm波长处都有吸收,栀子苷和芍药苷有最大吸收波长,而黄芩苷和大黄素的吸收很小,几乎测不到,故而本实验测定过程中选择双波长的检测模式,栀子苷和芍药苷在240 nm波长处有最大吸收波长,而黄芩苷和大黄素在275 nm波长处均有最大吸收波长,在该色谱条件下,色谱峰不拖尾,分离度好,阴性无干扰。
3.3 提取溶剂、提取方法及提取时间的选择 提取溶剂和处理方法对防风通圣丸(水丸)中栀子苷、芍药苷、黄芩苷和大黄素的含量测定有较大影响。在选择溶剂时用过甲醇,乙醇、甲醇和水的混合物,结果发现用甲醇提取时,所测物质的含量较高,其他成分干扰较少,所以本次实验制备样品溶液时采用甲醇进行提取。实验中还考察了30、40、50、60分钟超声提取法,对不同提取时间下的样品溶液分别进样比较发现,40、50分钟超声提取后所测的栀子苷、芍药苷、黄芩苷和大黄素的量基本相同,为了节约时间和减少杂质的含量,选择40分钟作为提取时间。
[1]国家药典委员会.中华人民共和国药典[M].北京:中国医药科技出版社,2010:96-616.
[2]黄山君,杨琪伟,石燕红,等.一测多评法测定白芍中芍药苷与芍药内酯苷的含量[J].中国中药杂志,2011,36(6):780-783.
[3]李姝梅,阳敬,熊磊,等.高效液相色谱法测定柴藿颗粒中黄芩苷的含量[J].儿科药学杂志,2012,18(3):43-45.
[4]肖林林,温建东,吴坚.RP-H PLC法测定小柴胡汤丸中黄芩苷的含量[J].中国现代药物应用,2011,5(22):66-67.
[5]张广春,陈明明.高效液相色谱法测定安神养心丸中五味子醇甲的含量[J].时珍国医国药,2011,22(7):1783-1784.
Contents of Jasminoidin,Peoniflorin,Baicalin and Emodin in FangFeng TongSheng Pills Determined by RP-HPLC Method
YANG Xiaojun1,FANG Bowen1,DING Yonghui2
1 Chemical Engineering Institute of Northwest University for Nationalities,Lanzhou 730124,China;2 Gansu Food and Drug Administration
Objective:To establish the determination methods for the contents of jasminoidin,peoniflorin,baicalin and emodin in FangFeng TongSheng pills simultaneously by RP-HPLC.Methods:The contents were determined under different wavelengths by adopting RP-HPLC as gradient elution.Chromatographic column:Zorbax E-clipse XDB-C18bonded silica gel column(4.6 mm×150 mm,5 μm);column temperature was room temperature;mobile phase:acetonitrile-2%phosphoric acid aqueous solution,gradient elution was adopted;flow rate 1.0mL/min;detection wavelengths were 240nm(jasminoidin and peoniflorin)and 275nm(baicalin and emodin)respectively.Results:Jasminoidin,peoniflorin,baicalin and emodin were in better linear relationship when their sample sizes were between 0.056 and 0.56g(r=0.999 7),0.103 5 and 1.029 7g(r=0.999 2),0.207 4 and 3.128 g(r=0.999 3),0.04 and 0.423 g(r=0.999 6),average recovery rates were 99.14%,99.65%,99.03%and 99.16%;RSD was 1.08%,1.01%,0.85%and 0.86%respectively.Conclusion:The method,simple,rapid,sensitive,reproducible and of high precision and accurate results,could be used for content determination of jasminoidin,peoniflorin,baicalin and emodin in FangFeng TongSheng pills simultaneously.
RP-HPLC;FangFeng TongSheng pills;jasminoidin;peoniflorin;baicalin;emodin;content determination
R927
A
1004-6852(2016)09-0027-08
2016-02-20
国家自然科学基金项目(编号21402153)。
杨小军(1982—),男,硕士学位,讲师。研究方向:药物分析。
