烷基-β-D-吡喃木糖苷的合成及性能
2016-11-14旷娜伍桂龙陈朗秋夏殊李贞操陈国勇叶雪
旷娜,伍桂龙,陈朗秋,夏殊,李贞操,陈国勇,叶雪
烷基-β-D-吡喃木糖苷的合成及性能
旷娜,伍桂龙,陈朗秋,夏殊,李贞操,陈国勇,叶雪
(湘潭大学化学学院,环境友好化学与应用省部共建教育部重点实验室,湖南湘潭,411105)
采用三氯乙酰亚胺酯法,以D-木糖为原料,经全乙酰化、C1位选择性脱乙酰基,转化成三氯乙酰亚胺酯,以一系列醇为受体与之进行偶联反应及脱保护,选择性地合成9种不同碳链长度的烷基-β-D-吡喃木糖苷。目标化合物经核磁共振技术对结构进行表征,利用偏光显微镜、热分析仪等对其性能进行测试。研究结果表明:正辛基和正壬基-β-D-吡喃木糖苷在低浓度时能使表面张力下降到较低值,且具有较好的发泡性,正壬基-β-D-吡喃木糖苷有较好的乳化性;当烷基链长≤9时,烷基β吡喃木糖苷的溶解熵都随温度升高呈下降趋势;当为7和8时,其糖苷具有较大的溶解焓;目标化合物均具有液晶现象。
木糖;烷基吡喃木糖苷;发泡;乳化;表面张力
糖类化合物及其衍生物不仅为生物体的生命活动提供必要的能量[1],而且在核酸、蛋白质、糖类化合物、脂质四大类生命分子中地位独特,起着联系核酸、蛋白质和脂质生物分子的枢纽作用。糖类化合物作为生物缀合物中结构各异的亲水性结构砌块,与蛋白质和脂类化合物等结合成糖蛋白和糖脂等影响着它们的折叠和功能响应[2−3]。木糖是被子植物细胞壁成分中含量居第2的以木聚糖(xylan)方式存在的最丰富的糖[4]。木聚糖属于杂多糖,由1,4-β-D-木糖残基组成主链,侧链上连有α-L-阿拉伯糖等,在农业残留物玉米的穗轴、棉籽壳等中质量分数较高[5],易于通过酸解法高效生产相应的木糖。糖类化合物廉价及丰富,羟基众多,具有优异的亲水性,通过结构改造,在其还原端与脂肪链的醇相连,形成高表面活性的双亲类型O-糖苷分子,符合表面活性剂的结构要求,从而可开发成糖苷类表面活性剂[6−7]。由于这种糖苷所用的主要原料为糖类化合物和脂肪链的醇,具有可再生性,在结构生物学、医学、洗涤剂、环保等领域具有广阔的应用价值。基于糖苷具有来源丰富、生态相容性好、配伍性良好、无毒性、稳定酶在水溶液中的催化功能作用,符合可持续发展要求,成为新型的表面活性剂[6−9]。烷基-β-D-吡喃木糖苷可采用酶催化法或化学合成法合成。尽管酶催化法选择性好、收率高、制备条件温和,但存在酶的筛选、活性的差别、溶剂的筛选、酶促反应的平衡与时间监控及复杂的分离纯化等问 题[9−13]。采用一锅法,以酸为催化剂使D-木糖或木聚糖与醇发生Fischer糖苷化反应,存在操作温度较高和反应时间较长、产物构型非单一、精制困难等问题;转糖苷法得到的也为精制困难的混合型糖苷;离子液体的引入虽然使反应效率提高,但存在离子液体污染产品等问题[14]。Koenigs−Knorr法为一种经典的立体选择性合成糖苷的方法,溴代木糖稳定性较差,特别容易分解,且主要采用银盐和汞盐等重金属的盐类为偶联反应的催化剂,容易造成重金属废液累积,严重污染环境,并存在反应的有效性与保护基团的选择性问题[9, 15];虽然PETROVIĆ等[16]采用维生素B12为偶联反应的催化剂,但存在成本过高的问题。采用全乙酰化的木糖(2)在四氯化锡催化下与一系列脂肪醇发生偶联反应,对时间要严格控制,否则有部分转化成α-异构体,当时间过长时,还会形成以α-糖苷为主的产物和少量2-位脱保护的糖苷副产物[17]。参考烷基-β-D-葡萄糖苷等合成与性能研究成果[8−9,15,18−21],按照Scheme1所示的路线,以D-木糖(1)为原料通过乙酰化、脱1-位乙酰基、与三氯乙腈反应,得到相应的三氯乙酰亚胺酯(4)。采用易于构筑1,2-反式糖苷键的三氯乙酰亚胺酯法,以三氟化硼乙醚作糖苷化偶联反应的催化剂,使得具有邻基参与效应的三氯乙酰亚胺酯(4)与相应的脂肪醇反应及相应的脱乙酰基保护,合成一系列烷基-β-D-吡喃木糖苷,如图1所示。本文作者对其溶解性、表面张力、乳化性能、液晶性能、热稳定性等理化性质进行研究。
1 实验
1.1 试剂与仪器
仪器为:DM-LM-P型偏光显微镜(德国leiea公司);BRUKER-AVANCE-400型核磁共振仪(瑞士Bruker公司);X-4数字显示显微熔点测定仪(河南巩义市英峪仪器厂); TGAQ50型热分析仪(美国PE公司);DP-A精密数字压力温度计(南京桑力电子设备厂);TLC分析使用青岛海洋化工厂分厂的涂层厚度为0.20~0.25 mm的HF254型硅胶板。
试剂为:显色观察体积分数为30%的硫酸甲醇溶液;使用青岛海洋化工厂的0.075~0.150 μm硅胶进行柱色谱分离。所用试剂均为市售化学纯或分析纯,实验用水均为蒸馏水。
1.2 正辛基-β-D-吡喃木糖苷(6e)的合成
往100 mL的圆底烧瓶中依次加入2.20 g (5.23 mmol)化学物4(见图1)[22]、10 mL二氯甲烷(4×10−10 m分子筛干燥)和2.48 mL(15.69 mmol)正辛醇,搅拌下冰水浴冷却至0 ℃左右,滴加0.65 mL (5.23 mmol)三氟化硼乙醚溶液,搅拌反应5 h,TLC(石油醚与乙酸乙酯体积比即(石油醚):(乙酸乙酯)=2:1)监测反应完全。反应液依次经饱和碳酸氢钠水溶液和饱和食盐水溶液洗涤,采用无水硫酸钠干燥,过滤,滤液浓缩,柱层析((石油醚):(乙酸乙酯)=10:1)分离,得到白色固体化合物5e(1.12 g,产率55%),用于下一步脱保护反应。

图1 6a~6i目标化合物的合成路线
往100 mL的圆底烧瓶中加入5.00 g (12.87 mmol)化合物5e和40 mL无水甲醇,搅拌溶解,用甲醇钠的甲醇稀溶液(甲醇钠质量与甲醇稀体积为1:5 g/mL)调节溶液的pH至约10.0,室温反应5 h,TLC(乙酸乙酯)监测反应完全。用732型阳离子树脂调节反应液的pH=7,过滤,滤液浓缩,柱层析(乙酸乙酯)分离,得到白色固体化合物6e(2.74 g,产率81%)。
其他烷基吡喃木糖苷(化合物6a~6d和6f~6i)的合成,分别用相应的醇代替,合成方法同上。
1.3 溶解性能测试
参考文献[8],在25 ℃测定目标产物6a~6f在不同溶剂中的溶解性。具体测定步骤如下:1) 在电子天平上准确称取定量的目标产物,放入测试杯中;2) 适当加入预定量溶剂,置于摇床上连续震摇1.0 h,使其在溶剂中达到溶解平衡;3) 根据其溶解情况,适量补加准确计量的溶剂,放入摇床上继续震摇,若未溶解的较少,则改为滴加;4) 观察溶解情况,当看不到溶质颗粒或液滴时,即认为全部溶解;5) 根据消耗溶剂的总量,计算得到其在该温度下的溶解度。
1.4 溶解焓的测定
参考文献[21−22] ,分别测出化合物6a~6f在15,25,35,45和55 ℃时溶解度3,再根据式(1)计算各烷基木糖苷的溶解焓(solH)。
由Gibbs-Duhem公式推导出:
则溶解熵(solS)为
1.5 乳化性能测试
参考文献[8],用移液管分别准确量取20 mL质量分数为0.12%的烷基-β-D-吡喃木糖苷水溶液和20 mL苯于100 mL具塞量筒中,盖好瓶塞,充分混合均匀后静置1.0 h,观察记录乳液层的体积(eb)和水层体积(wb)。按同样的方法测定烷基-β-D-吡喃木糖苷对菜籽油的乳化性能,观察记录乳液层体积(ez)和水层体积(wz)。
1.6 起泡力和泡沫的稳定性测定
参考文献[8],配制质量分数为0.12%的烷基-β-D-吡喃木糖苷水溶液100 mL,用移液管准确量取10.0 mL于100 mL具塞量筒中,盖好瓶塞,然后上下剧烈震荡1 min,立即测量泡沫的高度0,5 min之后再次测量泡沫的高度5。按下式计算泡沫消失速度,用来评价泡沫的稳定性:
=(0−5)/(60×5) (5)
显然,0越大,则发泡能力越强;越小,则泡沫的消失速度越慢,泡沫的稳定性越强。
1.7 表面张力的测定
参考文献[23],采用最大泡压法,配制一系列不同质量分数的烷基吡喃木糖苷(6c~6f)水溶液各25 mL,分别测出其在25 ℃时的最大附加压力,然后计算不同质量分数下的烷基木糖苷的表面张力。
1.8 热稳定性测试
在TGA(热分析仪)上测试烷基吡喃木糖苷的热稳定性。以N2作为保护气体,以20 ℃/min的速度升温,根据所获得的热质量损失曲线分析判定热分解温度。
1.9 热致液晶性观测
利用DM-LM-P型偏光显微镜,以3 ℃/min的升温速率观测烷基-β-D-吡喃木糖苷的热致液晶特性。
2 结果与讨论
2.1 烷基-β-D-吡喃木糖苷合成方法、1H NMR谱特征及其构型确认
在参考文献[16,18]的基础上,直接采用全乙酰化的木糖(2)在三氟化硼乙醚催化下与一系列脂肪醇发生偶联反应,TLC跟踪反应进程,发现出现许多分解的烤点,分离纯化负荷太大,产率较低,实际应用价值不高。按图1所示设计路线,通过保护、偶联与脱保护等糖化学策略,有效完成各种烷基-β-D-吡喃木糖苷(6a~6i)的合成。
吡喃木糖基三氯乙酰亚胺酯(4)与醇发生偶联反应得到烷基-2,3,4-三-O-乙酰基-β-D-吡喃木糖苷(5a~5i),后者通过脱乙酰基保护,获得相应的各种烷基-β-D-吡喃木糖苷(6a~6i),其熔点、1H NMR结果见表1。从表1可见:各种烷基-β-D-吡喃木糖苷1H NMR的H-1化学位移和耦合常数分别为6a(δ 4.40 (d,1,2= 5.3 Hz)),6b(δ 4.29 (d,1,2= 7.7 Hz)),6c(δ 4.31 (d,1,2= 6.5 Hz)),6d(δ 4.29 (d,1,2= 6.8 Hz)),6e(δ 4.33 (d,1,2= 6.2 Hz)),6f(δ 4.34 (d,1,2= 6.3 Hz)),6g(δ 4.36 (d,1,2= 5.9 Hz)),6h(δ 4.34 (d,1,2= 6.2 Hz)),6i(δ 4.07 (d,1,2= 7.4 Hz))。其1H NMR的H-1化学位移均在4.07~4.40之间,耦合常数1,2在5.3~7.7 Hz范围内,说明所合成的烷基-β-D-吡喃木糖苷6a~6i的糖苷键均为1,2反式的β-糖苷键[16−17]。

表1 烷基-β-D-吡喃木糖苷的熔点和1H NMR结果
2.2 烷基-β-D-吡喃木糖苷的溶解性
在室温条件下,合成的烷基-β-D-吡喃木糖苷(6a~6i)在水、甲醇、乙醇和乙酸乙酯中的溶解度(1)如图2所示。从图2可见:当烷基链碳数≥6时,木糖苷在水中的溶解度较低;当烷基链碳数>10时,基本不溶于水;在甲醇和乙醇中,木糖苷随其烷基链的增长溶解度逐渐下降;而在乙酸乙酯中,木糖苷的溶解度均较低。
为此,进一步测定烷基-β-D-吡喃木糖苷(6a,6c~6h)在各种醇(甲醇、乙醇、正丙醇、正丁醇、正戊醇、正己醇)中溶解度(2),结果见图3。从图3可见:在室温条件下,随着烷基链长度增长,木糖苷在同一种醇中的溶解度逐渐下降,而同一种烷基木糖苷的溶解度也随着醇的碳数的增长逐渐下降。所以,可以利用乙醇等短链醇(C1~C6)与水不同配比来增大木糖苷的溶解度。
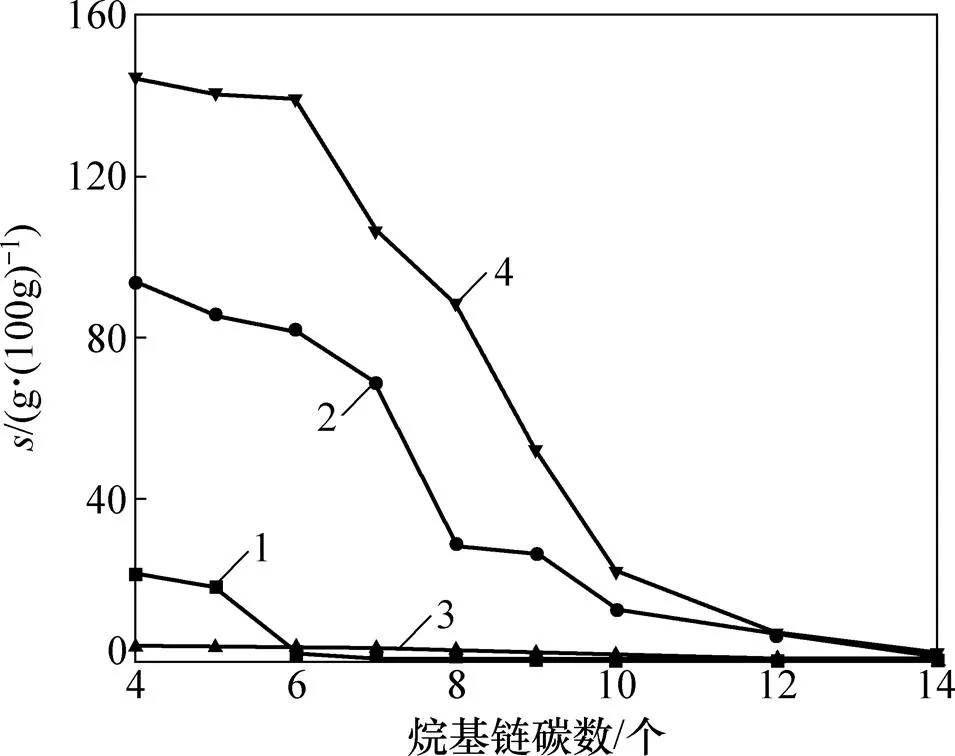
1—水;2—甲醇;3—乙醇;4—乙酸乙酯。
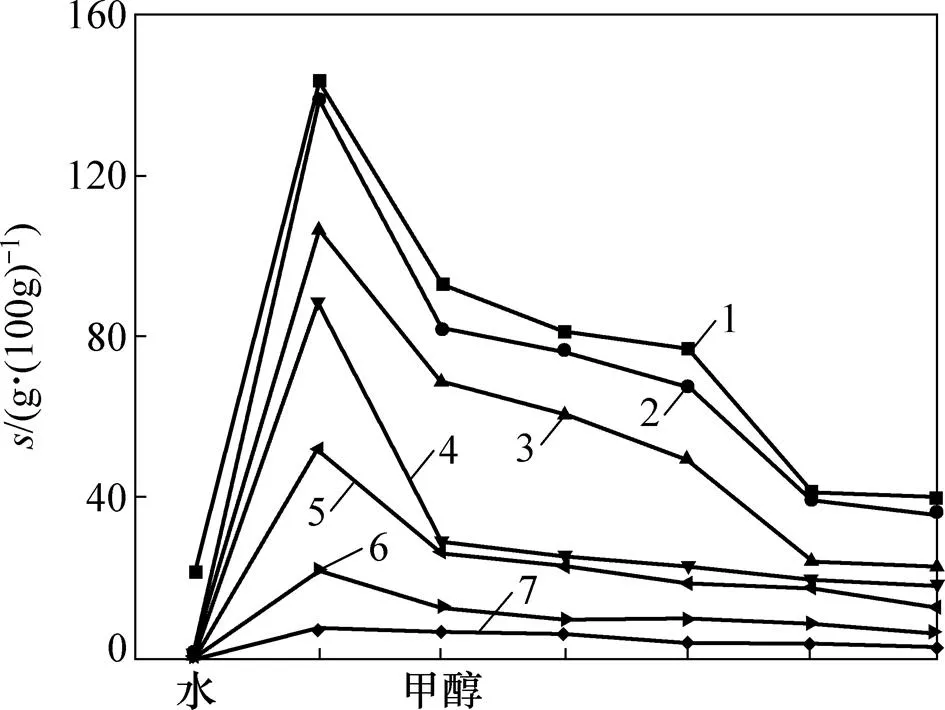
烷基木糖苷:1—6a;2—6c;3—6d;4—6e;5—6f;6—6g;7—6h。
2.3 烷基-β-D-吡喃木糖苷的溶解焓
在同一温度下,烷基-β-D-吡喃木糖苷随着具有憎水作用的烷基链长度的增长,亲水性逐渐变弱,表现为在水中的溶解度逐渐变小。在不同温度下,随着温度升高,同一烷基木糖苷溶解度增大。为了测定烷基木糖苷的溶解焓和溶解熵,分别选取室温下在水中有一定溶解度的烷基-β-D-吡喃木糖苷6a~6f测出在15,25,35,45和55 ℃下的溶解度,根据式(1)所示的温度和溶解度关系式(其中为热力学温度,为摩尔气体常数,solH为溶解焓),从图4可得到不同温度下溶解度曲线斜率(−solH/(2.30)),从而计算出烷基-β-D-吡喃木糖苷6a~6f的溶解焓(表2);根据式(4)绘出相应的溶解熵(solS)与温度的关系曲线,如图5所示。
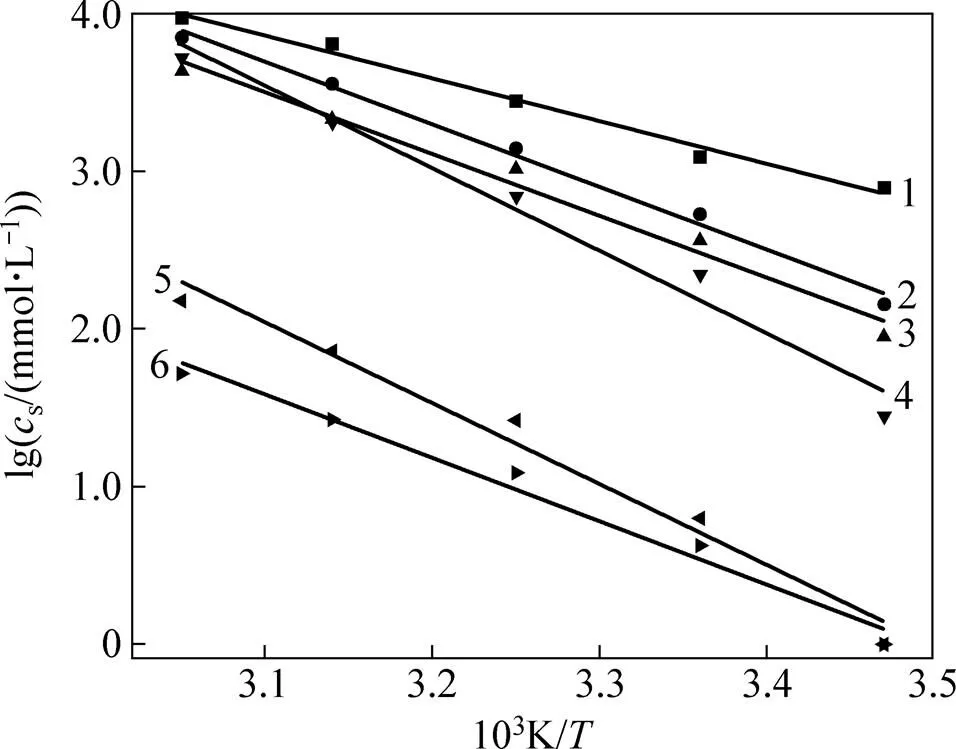
烷基木糖苷:1—6a;2—6b;3—6c;4—6d;5—6e;6—6f。

烷基木糖苷:1—6a;2—6b;3—6c;4—6d;5—6e;6—6f。

表2 木糖苷6a~6f的溶解焓
溶解焓为正值意味着糖苷溶于水的溶解过程是一个耗能过程。实质上,溶解是一种熵增加的过程,即糖苷溶于水时破坏溶剂水分子体系固有的次序,使水分子之间强的氢键作用削弱,但糖苷与水分子之间包括氢键在内的各种作用力,这种熵驱使的溶解作用所放出的能量不足以弥补溶剂水分子之间作用力削弱所需要的能量,所以,温度提高所提供的能量有助于糖苷在溶剂水中的溶解能力增强。从图5可见:各糖苷在水中溶解时其溶解熵呈现随温度上升而下降的线性关系,其中丁基-β-D-吡喃木糖苷6a的溶解熵最低,当碳链从4个(化合物6a)增长到7个和8个即庚基-β-D-吡喃木糖苷6d和辛基-β-D-吡喃木糖苷6e时溶解熵达到最大,随后有所下降(化合物6f)。
2.4 烷基-β-D-吡喃木糖苷的乳化性
烷基-β-D-吡喃木糖苷的表面性能与其烷基链的长度有关。图6所示为化合物6a~6f对菜籽油和苯的乳化性能。从图6可以看出:化合物6a~6f对苯和菜籽油乳化后,静置1 h后析出的水层体积(wb和wz)基本随烷基链的增长而减少,但基本相差不大,即木糖苷6a~6f对苯和菜籽油的乳化能力随烷基链的增长有略微增强;在=9时(化合物6f),对苯和菜籽油的乳化能力都达到最大。可能是由于壬基--吡喃木糖苷(6f)的烷基链较长,与疏水性有机化合物的作用能力强,从而使其与乳化物质(苯和菜籽油)作用形成界面膜的强度相应增加,乳状液液珠聚结时受到阻力增大,形成乳状液的稳定性提高,从而能够形成更加稳定的乳液层。

1—Vez;2—Vwz;3—Veb;4—Vwb。
2.5 烷基-β-D-吡喃木糖苷的起泡力及泡沫稳定性
木糖苷(6a~6f)的起泡性能见图7。图7表明:当木糖苷的烷基链长≥6(化合物6c~6f)时,具有良好的起泡性;随着烷基链长的增加,起泡性也逐渐增强;在=8时,起泡性最好,随后又减小;当=9时,壬基-β-D-吡喃木糖苷(6f)在低质量分数下(0.12%)具有很强的发泡力,且泡沫细腻。泡沫稳定性()随烷基链长的增加呈现出先下降后上升的趋势,当为8和9时最强。因为辛基、壬基木糖苷(=8,9)的亲水性基团与疏水性基团能够良好地匹配,在液体表面形成的液膜强度比较大,稳泡能力最强。

1—H0;2—H5;3—v。

烷基木糖苷:1—6c;2—6d;3—6e;4—6f。
2.6 烷基-β-D-吡喃木糖苷的表面张力
烷基-β-D-吡喃木糖苷的表面活性可以用其溶液降低表面张力的能力或效率来衡量,前者用表面活性剂使溶剂表面张力降低程度来衡量,后者用使表面张力降至一定值时所需要的表面活性剂质量分数来衡量。通过上述乳化性能和起泡性能测试结果可以看出:木糖苷6a,6b的表面活性较差,糖苷6c~6f具有良好的表面活性。本文采用最大泡压法,在25 ℃下,测定并计算出不同质量分数下烷基-β-D-吡喃木糖苷6c~6f所对应的表面张力。从图8可以得出:1) 几种烷基-β-D-吡喃木糖苷(6c~6f)添加到水中明显地降低了水的表面张力,呈现出两亲结构特征的木糖苷分子固有的表面活性;2) 在所测的质量分数范围内,烷基木糖苷(6c~6f)表面张力都有随质量分数增大先急剧下降而后又缓慢下降,最后基本不变的趋势;3) 不同的木糖苷6c~6f临界质量分数(CMC)不同。
表3所示为烷基-β-D-吡喃木糖苷6c~6f的临界质量分数及所对应的表面张力。溶液表面的吸附量达到饱和时(刚好饱和时的浓度即为临界浓度),表面张力最小。从表3可以看出:辛基-β-D-吡喃木糖苷(6e)达到临界浓度时对应的表面张力是最低的,能力最强。因为烷基木糖苷的表面活性是由其亲水糖基和疏水烷基链共同决定,当烷基链长为=8(糖苷6e)时,亲水性和疏水性达到最佳平衡,表面张力最低。
表4所示为糖苷6c~6f在同一浓度时对应的表面张力。从表4可见:当糖苷6c~6f质量浓度都为0.3 g/L时,表面张力随着烷基链长的增大而减小,糖苷6f使表面张力下降最低,效率最高。其原因可能是糖苷的烷基链越长,疏水性越强,所得糖苷逃离溶液内部而富集于溶液表面的倾向增大,因而其表面活性表现出随着烷基碳链长度的增加而增大的趋势。
2.7 烷基-β-D-吡喃木糖苷的热稳定性
图9所示为热质量损失结果。从图9可见:烷基吡喃木糖苷6a~6i 均只有1个质量损失台阶,起始分解温度分别为157.9,177.3,172.6,162.3,164.1,169.5,151.7,170.4和208.9 ℃左右,最大分解速率时的温度依次为283.2,287.8,296.6,257.9,290.4,286.1,301.9,262.7和339.8 ℃。糖苷6a~6i 热分解完时的温度分别为303.1,298.1,329.6,365.5,320.4,340.2,332.0,374.6和356.2 ℃。总质量损失率为94.9%,89.9%,91.0%,91.9%,95.3%,96.6%,89.6%,88.5%和91.5%。结果表明:合成的烷基吡喃木糖苷在150 ℃以下是稳定的。

表3 烷基-β-D-吡喃木糖苷6c~6f的临界胶束质量分数及其对应的表面张力

表4 烷基-β-D-吡喃木糖苷6c~6f质量浓度为0.3 g/L时对应的表面张力
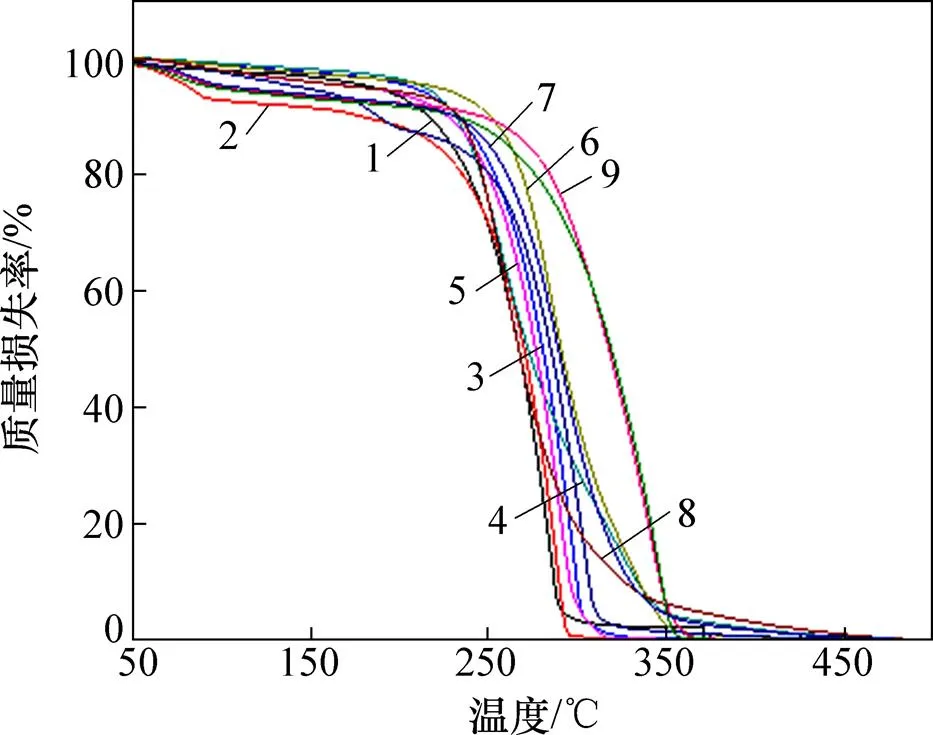
烷基木糖苷:1—6a;2—6b;3—6c;4—6d;5—6e;6—6f;7—6g;8—6h;9—6i。
2.8 烷基-β-D-吡喃木糖苷的热致液晶性
作为“软物质”(soft matter)的生物脂类通过自组装和自组织过程形成超分子液晶特征的动态变化的生物膜发挥其生物功能作用,葡萄糖脑苷脂等许多具有液晶性能的天然糖脂作为细胞膜的组分与生物活性和疾病的成因有关[9,19,24]。本研究利用偏光显微镜直接考察不同链长的烷基-β-D-吡喃木糖苷6a~6i的热致液晶行为[8]。当烷基木糖苷处于液晶相时,可依赖其分子间相互作用采用自组装和自组织模式形成氢键强作用的糖环层和憎水弱作用的烷基链层依次相间排列,自发组装成层状、条纹、镶嵌、平行排列的织构。
具有液晶特征的化合物因加热而呈现所谓的“双熔融转变”的相变现象[9],当加热达到其熔点(p)时,结晶性的固态首先转变成半透明的液晶态,接着在较高的温度(p)下转变成各向同性的清亮的液态。表5所示为木糖苷的相转变温度。从表5可见:烷基-β-D-吡喃木糖苷属于一头一尾(one head one tail)或一头组一链(one head group-one chain)且呈现多态性的双亲吡喃糖苷分子[19],在加热时其相变发生在相对较小的温度区间,但都出现了液晶相,且当疏水性烷基碳链长度为6时,烷基-β-D-吡喃木糖苷相变温度之差(Δ)较大,其他的都比较接近。在木糖苷中,烷基有助于提升分子取向的稳定性,且这种稳定性对液晶相的生成是必要的。但由于烷基-β-D-吡喃木糖苷为环状的糖苷只有3个游离羟基,比烷基--D-吡喃葡萄糖苷少了亲水性头部的糖环上的羟甲基,也比木糖醇的十二烷基单醚少了1个游离的羟基,清亮点p不高,所以,得到的烷基-β-D-吡喃木糖苷具有液晶相,但相变温度范围(∆=p−p)较窄,液晶相的稳定性较弱。
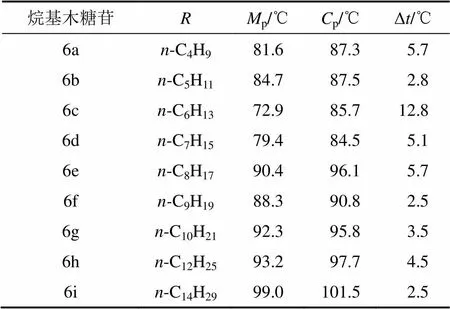
表5 木糖苷的相转变温度
3 结论
1) 三氯乙酰亚胺酯法具有高度立体选择性、克服了溴代糖法存在的重金属污染的弊端,采用此法,以木糖为原料,通过全乙酰化、选择性1-位脱保护、转变成三氯乙酰亚胺酯、偶联、脱保护共计5步反应,合成了9种烷基-β-D-吡喃木糖苷(6a~6i),并系统地测定了其溶解性、溶解焓、表面张力、乳化性、发泡力、泡沫稳定性、热稳定性及热致液晶性。
2) 所合成的烷基-β-D-吡喃木糖苷(6a~6i)在水中的溶解性比较好,且糖苷6d(=7)和糖苷6e(=8)的溶解焓最大。糖苷6e(=8)和6f (=9)能使表面张力下降到较低值,表面活性较强,糖苷6e(=8)使表面张力降低到最低值,即能力最强;糖苷6f (=9)使表面张力下降到一定值时所需的浓度最小,即效率最高。糖苷6f (=9)对苯和菜籽油的乳化能力最强。糖苷6e(=8) 和6f(=9)的起泡力和泡沫稳定性均较好。
3) 所合成的烷基-β-D-吡喃木糖苷(6a~6i)在150 ℃以下是稳定的,且液晶相变温度范围较窄。其有关应用、细胞毒性与结构改造等构效关系有待于进一步研究。
参考文献:
[1] HENDERSON P J F, BALOWIN S A. Bundles of insights into sugar transporters[J]. Nature, 2012, 490(7420): 348−350.
[2] TAYLOR M E, DRICKAMER K. Convergent and divergent mechanisms of sugar recognition across kingdoms[J]. Current Opinion in Structural Biology, 2014, 28: 14−22.
[3] NAGAE M, YAMAGUCHI Y. Three-dimensional structural aspects of protein–polysaccharide interactions[J]. International Journal of Molecular Sciences, 2014, 15(3): 3768−3783.
[4] SATO T K, LIU T, PARREIRAS L S, et al. Harnessing genetic diversity in Saccharomyces cerevisiae for fermentation of xylose in hydrolysates of alkaline hydrogen peroxide-pretreated biomass[J]. Applied and Environmental Microbiology, 2014, 80(2): 540−554.
[5] CORREIA M A S, MAZUMDER K, BRÁS J L A, et al. Structure and function of an arabinoxylan-specific xylanase[J]. The Journal of Biological Chemistry, 2011, 286(25): 22510−22520.
[6] ZAHID N I, CONN C E, BROOKS N J, et al. Investigation of the effect of sugar stereochemistry on biologically relevant lyotropic phases from branched-chain synthetic glycolipids by small-angle X-ray scattering[J]. Langmuir, 2013, 29(51): 15794−15804.
[7] CHAI Jinling, WU Changju, LI Ganzuo, et al. Studies on middle-phase microemulsions of green surfactant n-dodecyl polyglucoside C12G1.46[J]. Chinese Journal of Chemistry, 2003, 21(1): 25−29.
[8] 刘灯峰, 陈朗秋, 李宏伟, 等. 烷基-β-D-吡喃葡萄糖苷的合成与性能[J]. 应用化学, 2013, 30(10): 1120−1126. LIU Dengfeng, CHEN Langqiu, LI Hongwei, et al. Syntheses and properties of alkyl β-D-glucopyranosides[J]. Chinese Journal of Applied Chemistry, 2013, 30(10): 1120−1126.
[9] XU W, OSEI-PREMPEH G, LEMA C, et al. Synthesis, thermal properties, and cytotoxicity evaluation of hydrocarbon and fluorocarbon alkyl β-D-xylopyranoside surfactants[J]. Carbohydrate Research, 2012, 349: 12−23.
[10] MATSUMURA S, SAKIYAMA K, TOSHIMA K.Preparation of octyl β-D-xylobioside and xyloside by xylanase-catalyzed direct transglycosylation reaction of xylan and octanol[J]. Biotechnology Letters, 1999, 21(1): 17−22.
[11] KADI N, BELLOY L, CHALIER P, et al. Enzymatic synthesis of aroma compound xylosides using transfer reaction by Trichoderma longibrachiatum xylanase[J]. Journal of Agricultural and Food Chemistry, 2002, 50(20): 5552−5557.
[12] LI Y K, YAO H Y, CHO Y T. Effective induction, purification and characterization of Trichoderma koningii G-39 β-xylosidase with high transferase activity[J]. Biotechnology and Applied Biochemistry, 2000, 31(2): 119−125.
[13] SHINOYAMA H, KAMIYAMA Y, YASUI T. Enzymatic synthesis of alkyl β-xylosides from xylobiose by application of the transxylosyl reaction of Aspergillus niger β-xylosidase[J]. Agricultural and Biological Chemistry, 1988, 52(9): 2197−2202.
[14] SEKINE M, KIMURA T, KATAYAMA Y, et al. The direct and one-pot transformation of xylan into the biodegradable surfactants,alkyl xylosides, is aided by an ionic liquid[J]. RSC Advances, 2013, 3: 19756−19759.
[15] SATGÉ C, BRAS J L, HÉNIN F, et al. DMF promoted xylosylation of terpenols[J]. Tetrahedron, 2005, 61(35): 8405−8409.
[16] PETROVIĆ Z D, ANDJELKOVIĆ D, SPASOJEVIĆ A. Vitamin B12and BF3-etherate as catalysts in synthesis of some C4-C12-alky β-D-xylopyranosides[J]. Indian Journal of Chemistry, 2006, 45B: 272−275.
[17] KONSTANTINOVIĆ S, PETROVIĆ Z, SPASOJEVIĆ A, et al. Synthesis of C7-C16-alkyl glycoside: part II. synthesis of alkyl D-xylopyranosides in the presence of tin(IV) chloride as a Lewis acid catalyst[J]. Indian Journal of Chemistry, 2001, 40B(7): 614−618.
[18] OLDHAM E D, SEELAM S, LEMA C, et al. Synthesis, surface properties, and biocompatibility of 1,2,3-triazole-containing alkyl β-D-xylopyranoside surfactants[J]. Carbohydrate Research, 2013, 379: 68−77.
[19] GOODBY J W, GÖRTZ V, COWLING S J, et al. Thermotropic liquid crystalline glycolipids[J]. Chemical Society Reviews, 2007, 36(12): 1971−2032.
[20] 蔡水根, 陶冠军, 秦昉, 等. L-抗坏血酸月桂酸酯的酶法合成、分离及其性质[J]. 食品工业科技, 2008, 29(10): 211−215. CAI Shuigen, TAO Guanjun, QIN Fang, et al. Enzymatic synthesis, separation and properties of L-ascorbyl laurate[J]. Science and Technology of Food Industry, 2008, 29(10): 211−215.
[21] LI Yu, XU Li, WANG Fuan, et al. Solubilities of cefepime hydrochloride in water + (ethanol, 1-propanol, or 2-propanol) from (278.15 to 308.15)K[J]. Journal of Chemical & Engineering Data, 2010, 55(9): 4098−4103.
[22] MANI K, BELTING M, ELLERVIK U, et al. Tumor attenuation by 2(6-hydroxynaphthyl)-β-D- xylopyranoside requires priming of heparan sulfate and nuclear targeting of the products[J]. Glycobiology, 2004, 14(5): 387−397.
[23] 邓月娥, 宋捷, 龚文君, 等. 十二烷基多苷的性能研究[J]. 应用化工, 2010, 39(8): 1164−1166. DENG Yuee, SONG Jie, GONG Wenjun, et al. Study on the properties of dodecyl polyglucosides[J]. Applied Chemical Industry, 2010, 39(8): 1164−1166.
[24] MARTIN P, GODÉ P, VILLA P, et al. D-xylose derivative liquid crystals[J]. Journal of Thermal Analysis and Calorimetry, 2001, 63(2): 339−344.
(编辑 陈灿华)
Synthesis and properties of alkyl--xylopyranosides
KUANG Na, WU Guilong, CHEN Langqiu, XIA Shu, LI Zhencao, CHEN Guoyong, YE Xue
(Key Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education,College of Chemistry, Xiangtan University, Xiangtan 411105, China)
Using the trichloroacetimidate method and taking D-xylose as a raw material, nine kinds of carbon chain length alkyl β-D-xylopyranosides were synthesized selectively through acetylation, selective deacetylation at C1 position, and conversion to trichloroacetimidate, coupling with a series of acceptors alcohols and deprotection. The structures of target compounds were characterized by NMR technology, and their properties were tested by polarization microscopy(POM), thermal gravimetric analysis (TGA), etc. The results show that the n-octyl and n-nonyl β-D-xylopyranosides are able to make surface tension decrease to a low value and possess better foaming properties, while n-nonyl β-D-xylopyranoside has better emulsification. However, with the increase of temperature, dissolution entropies of all alkyl β-D-xylopyranosides decrease when the chain length of alkyl group is less than 9. The dissolution enthalpies of D-xylopyranosides are bigger when the chain length of alkyl groups are 7 and 8. All alkyl β-D-xylopyranosides can form thermotropic liquid.
xylose; alkyl β-D-xylopyranoside; surface tension; foaming; emulsification
10.11817/j.issn.1672-7207.2016.10.006
O629.11+3
A
1672−7207(2016)10−3323−09
2015−11−12;
2015−01−15
湖南省自然科学基金资助项目(14JJ2067,10JJ6023);湘潭大学第十批大学生创新基金资助项目(2014xtuxj31)(Projects(14JJ2067, 10JJ6023) supported by the Natural Science Foundation of Hunan Province; Project(2014xtuxj31) supported by the Tenth College Students Innovation Fund of Xiangtan University)
陈朗秋,教授,从事糖化学、有机合成、药物合成和食品添加剂研究;E-mail:chengood2003@263.net
