不同发酵时期酸马奶细菌群落结构
2016-11-12布仁其其格高雅罕任秀娟包艳青魏睿元赵一萍韩海格乌云达来黄金龙内蒙古农业大学动物科学学院内蒙古自治区蒙古马遗传资源保护及马产业工程实验室内蒙古呼和浩特0008内蒙古医科大学基础医学院内蒙古呼和浩特000
布仁其其格,高雅罕,任秀娟,包艳青,魏睿元,赵一萍,韩海格,乌云达来,黄金龙,芒 来,*(.内蒙古农业大学动物科学学院,内蒙古自治区蒙古马遗传资源保护及马产业工程实验室,内蒙古 呼和浩特 0008;2.内蒙古医科大学基础医学院,内蒙古 呼和浩特 000)
不同发酵时期酸马奶细菌群落结构
布仁其其格1,2,高雅罕1,任秀娟1,包艳青1,魏睿元1,赵一萍1,韩海格1,乌云达来1,黄金龙1,芒 来1,*
(1.内蒙古农业大学动物科学学院,内蒙古自治区蒙古马遗传资源保护及马产业工程实验室,内蒙古 呼和浩特 010018;2.内蒙古医科大学基础医学院,内蒙古 呼和浩特 010110)
以聚合酶链式反应扩增16S rRNA基因序列采用454焦磷酸测序方法分析酸马奶传统发酵过程中细菌群落结构演替变化。结果表明:在发酵的初期细菌多样性最高,而细菌丰度在72 h时最高;硬壁菌门(Firmicutes)和变形菌门(Proteobacteria)为酸马奶中的优势细菌门;乳杆菌 属(Lactobacillus)和乳球菌属(Lactococcus)为其优势细菌属;随着发酵时间的延长,各个细菌门与属都存在上升或下降的趋势变化。本研究可为其他乳制品发酵过程中细菌群落结构研究提供借鉴。
酸马奶;传统发酵;细菌群落结构;16S rRNA
布仁其其格, 高雅罕, 任秀娟, 等. 不同发酵时期酸马奶细菌群落结构[J]. 食品科学, 2016, 37(11): 108-113. DOI:10.7506/spkx1002-6630-201611019. http://www.spkx.net.cn
Burenqiqige, GAO Yahan, REN Xiujuan, et al. Dynamic changes of bacteria community structure during koumiss fermentation[J]. Food Science, 2016, 37(11): 108-113. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-201611019. http://www.spkx.net.cn
传统发酵酸马奶是以马奶为原材料与发酵剂混合,在平均20~23 ℃温度下定时上下捣拌一定次数,发酵2~3 d即可成为略带酸味、微喷酒香、沁人心脾、清爽适口的发酵乳饮品。发酵剂为上一个发酵周期保留的酸马奶或上一年度酸马奶通过直接冷冻保存或借助媒介(比如:蘸吸酸马奶的干净皮子、毡块、米、野果等)干燥保留物,其中含有复杂的微生物群落结构,长期连续参与到酸马奶发酵中对其品质起到及其重要的作用。
酸马奶发酵微生物中细菌是重要组成部分。1985年,研究已发现酸马奶中包含乳杆菌和乳球菌[1]。在新疆地区[2]、内蒙古锡林郭勒盟[3]以及蒙古国[4]采集的传统发酵酸马奶中分别分离培养出不同数量的乳杆菌菌株。Hao等[5]采用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)技术研究了酸马奶中细菌生物多样性,而刘芳等[6]采用16S rRNA基因测序技术分析了酸马奶乳酸菌菌群多样性。伴随分子生物学研究手段的引入对于酸马奶中细菌的研究已取得了一定成果。然而,基于某一固定时间点采样研究自然发酵酸马奶中细菌群落,不能全面、客观了解酸马奶发酵过程中细菌群落的真实存在状态和动态变化过程。2001年,殷文政等[7]采用传统方法对酸马奶传统发酵过程中细菌进行分类鉴定,初步了解群落结构动态变化情况。由于培养基方法的局限性[8]无法全面反映真实状况,因此必须运用相对更加精确的技术研究酸马奶发酵过程中细菌群落结构变化情况。
细菌群落结构及多样性研究中应用越来越多的方法是对所提取到的环境微生物宏基因组DNA进行16S rRNA基因(rDNA)序列的分析。16S rRNA基因全长为 1 542 bp,由 9 个可变区(V1~V9)和10 个保守区组成,保守区反映物种间的亲缘关系,而可变区则揭示物种间的差异,适用于各类细菌生物进化和系统分类学研究[9]。针对16S rRNA基因的保守区域设计引物,所得不同的序列代表不同的菌种,经测序分析便可确定群落结构和多样性。
近年来,迅速发展并推广应用的第2代高通量测序技术被应用到对16S rRNA基因的聚合酶链式反应(polymerase chain reaction,PCR)产物测序中,可以同时对数十万甚至数百万序列片段进行测序且互不干扰[10]。高通量测序平台中Roche公司454焦磷酸测序仪测序单序列读长长(目前升级后的GS FLX+最长可达到1 000 bp)、可重复性和精确性高,已被广泛应用于如海洋、湖泊、空气、土壤、矿井、发酵食品[11]、反刍动物瘤胃、人类口腔及肠道等多种环境样品的微生物群落结构多样性研究。
为了探究传统发酵过程中酸马奶细菌群落结构的动态变化过程,在内蒙古锡林郭勒盟镶黄旗选取当地酿制酸马奶时间最久,品质最具有代 表性的牧户,模拟其发酵酸马奶过程进行样品采集。后期对细菌16S rRNA基因序列进行454焦磷酸测序以及序列分析,从非培养水平上阐述酸马奶发酵过程中细菌种群多样性和群落结构动态变化规律,结果将对酸马奶传统发酵过程中微生物动态变化研究,酸马奶发酵机理研究,产品质量监控等具有实际的参考意义。
1 材料与方法
1.1 试剂
E.Z.N.A Soil DNA提取试剂盒 美国Omega公司;DL2000 DNA分子质量标准、TransStart Fastpfu DNA Polymerase、Taq DNA聚合酶、10×Loading Buffer、6×Loading Buffer 日本TaKaRa公司。
1.2 仪器与设备
Roche GS FLX 基因组测序仪 瑞士罗氏公司;UVP CDS-8000凝胶成像分析系统 美国Bio-Rad公司;Eppendorf 5810R台式高速冷冻离心机 德国Eppendorf公司;GeneAmp®9700型PCR仪 美国ABI公司;QuantiFluor™-ST蓝色荧光定量系统 美国Promega公司。
1.3 方法
1.3.1 样品
2013年7月,在内蒙古锡林郭勒盟镶黄旗,参照牧户家做法酿制酸马奶。发酵剂采用牧民家中酸马奶,鲜马奶与发酵剂以体积比13∶1混匀,一个体系3.5 L,共有3 个平行发酵(编号A、B、C),温度为(22±2) ℃。每隔2.5 h捣拌300 次,每日共5 次。收集0~96 h的样品,采样之前测定pH值,液氮保存样品带回实验室。
1.3.2 宏基因组DNA提取与检测
选取0、12、24、48、72、96 h等6 个时期样品,采用E.Z.N.A Soil DNA提取试剂盒按说明提取酸马奶宏基因组DNA。
1.3.3 PCR扩增16S rRNA 基因序列V1~V3区
20 μL反应体系:5×Fast Pfu Buffer 4 μL、2.5 mmol/L dNTPs 2 μL、Forward Primer(5 μmol/L)0.8 μL、Reverse Primer(5 μmol/L)0.8 μL、FastPfu Polymerase 0.4 μL、Template DNA 10 ng,补ddH2O至20 μL。
引物序列为:27F 5’-AGAGTTTGATCCTGGCTC AG-3’,533R 5’-TTACCGCGGCTGCTGGCAC-3’,测序起始端为533R,引物中加入7 个核苷酸标签(barcode)。PCR扩增条件为:95 ℃预变性2 min,95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,共27 个循环,最后72 ℃延伸5 min。
1.3.4 样品平衡及测序
PCR产物检测定量,按每个样品100 nmol/L的浓度将DNA样品(每个样品的核苷酸标签barcode不重复)混为一管,上机测序,由上海美吉生物公司完成。
1.3.5 高质量序列筛选
筛选原则:序列barcode和引物必须完整;序列的长度大于300 bp;整条序列上质量大于20的碱基所占比例须高于93%。
1.3.6 生物信息学处理
使用QIIME(v1.4.0)分析平台开展序列的生物信息学分析[12-13]。PyNAST校准排齐[14]序列,以100%相似性进行UCLUST归并从而建立无重复的533R→27F序列集。采用两步UCLUST归并,在100%相似性归并的基础上进一步进行97%相似性的归并,从而建立分类操作单元(operational taxonomic units,OTUs)。ChimeraSlayer[15]去除可能存在嵌合体序列的OTU,继而使用核糖体数据库项目(Ribosomal Database Project,RDP)工具classifi er[16]贝叶斯算法对OTU代表序列进行分类学鉴定。FastTree软件[17]生成OTU代表序列的系统发育进化树,在此基础上进行Alpha多样性计算。Alpha多样性包括超1指数(Chao1 index)和香农指数(Shannon index),分别对样品菌群构成的丰度和多样性进行评价。
香农指数计算公式为:

式中:Sobs为实际测量出的OUTs数目;ni为含有i 条序列的OTUs数目;N为所有的序列数。
超1指数计算公式为:
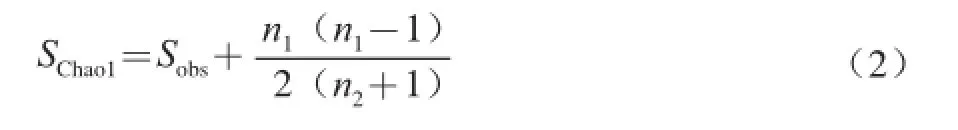
式中:SChao1为估计的OUTs数;Sobs为实际测量出的OUTs数;n1为只有一条序列的OTUs数目;n2为只有两条序列的OTUs数目。用于指数评估的OTUs相似水平97%。
同时使用稀疏曲线(rarefaction curve)和香农指数曲线评估每个样本测序的多样性以及在不同的OTUs划分水平下的多样性,以评价测序量是否能够代表原始群落的多样性。基于UniFrac[18]距离对样品之间进行加权(weighted)和非加权(unweighted)的主坐标分析(principal coordinate analysis,PCoA),同时采用基于UniFrac的非加权组平均法(unweighted pair-group method with arithmetic means,UPGMA)进行样品聚类。
1.3.7 数据的统计分析及图表绘制
基于Matlab R2011b(The MathWorks, Natick, MA, USA)软件,使用多元方差分析(multivariate analysis of variance,MANOVA)对不同时间点的酸马奶细菌菌群群落结构的差异进行显著性分析,作图采用Origin 8.0软件。
2 结果与分析
2.1 样品16S rRNA基因序列质量评估
通过454焦磷酸高通量测序,18 个样品共产生67 514 条高质量16S rRNA基因序列,每个样品平均产生3 750 条。经过PyNAST alignment和100%序列鉴定聚类分析后,共得到11 456 条代表性序列。继而根据序列的97%相似性进行OTU划分及嵌合体检查,得到477 个OTUs进行下一步分析。
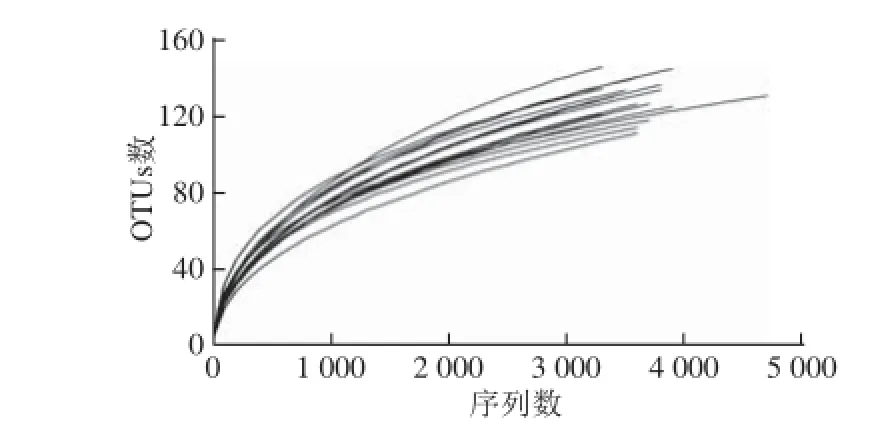
图1 稀疏曲线Fig. 1 Sparse curve

图2 香农指数图Fig. 2 Shannon index map
依据当前的测序量每个样品的稀疏曲线不能进入平台期(图1),但是Shannon多样性曲线已经饱和(图2),表明即使随着测序量的增加新的种系型可能会被发现,但是细菌多样性已经不再随之发生变化,现有测序量可以反映样品中绝大多数的细菌物种信息。
2.2 序列丰度和多样性分析
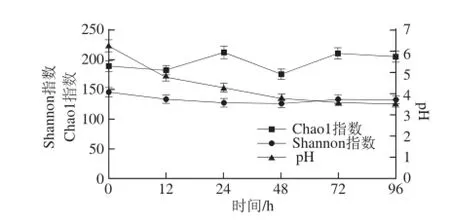
图3 酸马奶发酵过程中Chao1指数、Shannon指数和pH值变化Fig. 3 Changes in Chao1 index, Shannon index and pH level during the fermentation process of koumiss
以Chao1指数对不同发酵时间点酸马奶中细菌菌群丰度进行评价,如图3所示,显示出发酵过程中细菌的丰度具有明显的起伏变化。发酵起始12 h后细菌开始大量繁殖直至24 h,此后繁殖速率下降在48 h时细菌丰度最低,而72 h时转为丰度最高。由图3中Shannon指数变化情况可知,在酸马奶发酵过程中细菌多样性存在上下波动。0 h时样品中细菌的多样性较高,主要来源于发酵剂,随着发酵时间的延长多样性逐渐下降,而在72 h时其丰度会增至最高。
Chao1指数与Shannon指数变化规律表明在酸马奶发酵24 h之内细菌群落内部存在激烈的生存竞争,部分菌群取得生存优势而大量繁殖。而24 h之后的生存环境更加恶化,直至48 h之后pH值变化缓慢趋于稳定期,细菌丰度与多样性都在增高,细菌群落结构与营养供给[19]和环境酸度[20]有关。
2.3 基于门属水平的不同发酵时间点酸马奶细菌构成的研究
采用RDP数据库同源性序列比对与聚类相结合的方法对序列进行鉴定,产生了17 个门,其中含量最多的2 个细菌门占到了测序序列总数的98.91%,分别为硬壁菌门(Firmicutes,85.55%)和变形菌门(Proteobacteria,13.36%)。此外检测到拟杆菌门(Bacteroidetes,0.59%)、放线菌门(Actinobacteria,0.36%)、氰基细菌(Cyanobacteria,0.05%)和栖热菌门(Deinococcus-Thermus,0.001%)所占比例较低。新疆地区酸马奶研究[21]显示优势细菌门同样是硬壁菌门(95%以上),其次为变形菌门(低于5%),两个地区酸马奶发酵的优势细菌门相同,比例上存在差异。
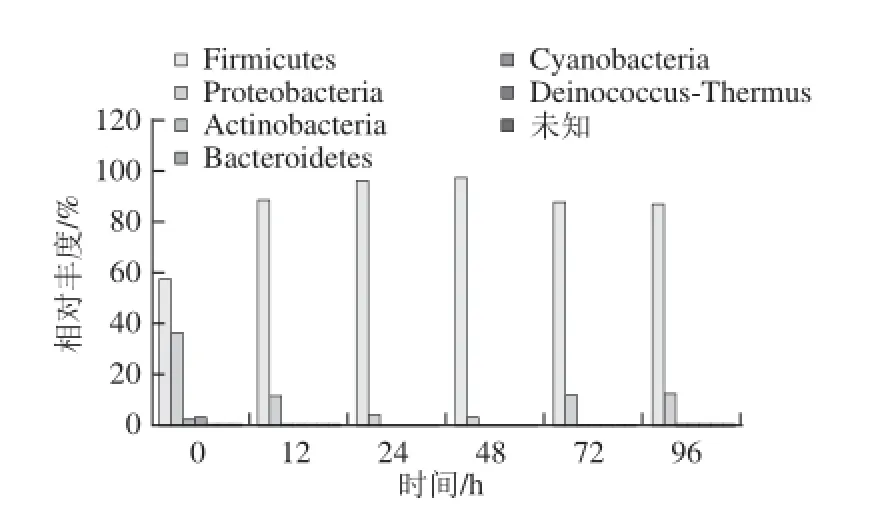
图4 基于门水平的不同发酵时间点酸马奶细菌丰度变化Fig. 4 Changes in bacterial abundance at phylum level during the fermentation process of koumiss
由图4可知,酸马奶发酵至48 h硬壁菌门含量呈现出上升的趋势,而后开始下降至72 h,随后趋于稳定。细菌群落结构中硬壁菌门在酸马奶发酵过程中一直占据主导地位,是酸马奶发酵的优势细菌门。变形菌门是酸马奶发酵的次优势细菌门,其序列数量变化呈现出与硬壁菌门相反的趋势,表明在发酵过程中两者之间存在相互作用。
经序列比对法对序列进行鉴定共得到54 个属,有0.25%的序列不能鉴定到属水平,表明酸马奶发酵过程中细菌具有丰富的多样性,还有发现新的菌种的可能性。分类结果显示在酸马奶传统制作过程中乳杆菌属(Lactobacillus)为优势细菌属,乳球菌属(Lactococcus)为次优势细菌属。殷文正等[7]在其研究中也发现酸马奶发酵过程中乳杆菌属为优势细菌属。酸马奶发酵过程中细菌在属水平上的多样性动态变化如图5所示,在发酵起始12 h后乳杆菌属的繁殖速率加快,此时环境中酸度值能够满足其启动生长,24 h后其含量又呈现出下降的趋势,可能是菌群大量繁殖后出现了养分竞争而抑制了繁殖速率。与此相比,发酵起始后酸马奶中乳球菌属繁殖速率较快,至12 h后随着乳杆菌属数量增多其繁殖速率开始下降,当乳杆菌属自24 h开始数量下降时其繁殖速率又升高。72 h后乳杆菌属数量增加,而乳球菌属数量减少,在发酵过程中似乎两者之间存在某种相互作用;酸马奶发酵起始所含醋酸菌属(Acetobacter)、不动杆菌属(Acinetobacter)等多个细菌属数量在发酵过程中减少,只有醋酸菌属在48 h后其数量明显增多;酸马奶发酵过程中检测到埃希菌属(Escherichia)和志贺菌属(Shigella)等致病菌,其平均含量为0.5%,发酵过程中受到抑菌活性物质[22]以及酸度变高[23]等影响数量越来越少。
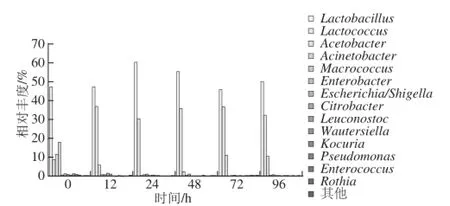
图5 基于属水平的不同发酵时间点酸马奶细菌丰度变化Fig. 5 Changes in bacterial abundance at genus level during the fermentation process of koumiss
2.4 基于多元统计方法的不同时间点酸马奶细菌群落结构的比较分析
基于UniFrac的加权和非加权主坐标分析,其第一主成分(PC1)和第二主成分(PC2)的贡献率分别为65.23%和17.80%、25.97%和10.32%。基于UniFrac的加权(图6)和非加权主坐标分析图(图7)结果基本一致,即0 h时3 组样品的细菌群落结构是相似的,随着发酵时间的延长,A、B两组样品呈现出一定的交叠现象,说明A、B两组样品的细菌群落结构呈现出一定的相似性。而C组样品呈现出一定的聚类趋势,说明C组样品相对于A、B两组样品来讲具有自己特殊的细菌群落结构。结果表明酸马奶传统发酵过程中,除了温度与捣拌次数以外还有其他因素容易影响到细菌群落结构而使其发生改变。
MANOVA基于Weighted Unifrac PCoA前85%主成分分析基础上的马斯距离聚类结果显示(图8),0 h酸马奶的细菌菌群群落结构与其他时间具有极显著差异(P<0.001)。12、72 h与96 h之间酸马奶细菌群落结构差异不显著(P>0.05),24 h与48 h之间细菌群落结构差异不显著(P>0.05),然而12、72、96 h与24、48 h之间酸马奶细菌群落结构差异极显著(P<0.001)。经过不同时间点酸马奶细菌群落结构的比较分析发现酸马奶发酵过程中细菌群落结构存在显著动态变化。细菌群落结构变化差异性将发酵过程大体划分为3 个阶段,即0~12、24~48、72~96 h。

图6 基于操作分类单元(9977%相似水平)相对含量的不同发酵时间点酸马奶细菌菌群结构Unifrac加权PCoA分析分布图Fig. 6 Principal coordinate analysis (PCoA) score plot based on the relative abundance of OTUs (97% similarity level) at different time points during the fermentation process of koumiss
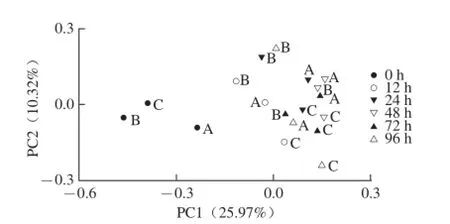
图7 基于操作分类单元(9977%相似水平)相对含量的不同发酵时间点酸马奶细菌菌群结构Unifrac非加权PCoA分析分布图Fig. 7 Principal coordinate analysis (PCoA) score plot based on the relative abundance of OTUs (97% similarity level) of bacterial community structure atdifferent time points during the fermentation process of koumiss
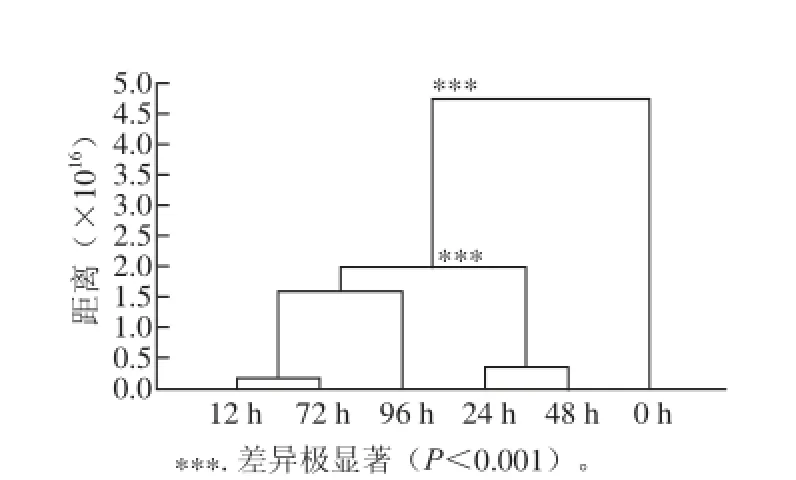
图8 基于MANOVA分析的不同发酵时间点酸马奶细菌群落结构马斯距离聚类分析Fig. 8 Mahalanobis distance cluster analysis based on MANOVA analysis of bacterial community structure at different time points during the fermentation process of koumiss
2.5 核心细菌群分析
研究发现不同的发酵时间点酸马奶中细菌菌群构成差异较大,但经过进一步分析发现它们具有相同的核心菌群。由18 个样品所得477 个OTUs中,有24 个OTUs存在于所有的样品,其中1 个OTU为巨型球菌属,9 个OTUs为乳球菌属,13 个OTUs为乳杆菌属,1个OTU为醋酸菌属,即在酸马奶发酵96 h过程中有4 个菌属一直存在,对于酸马奶品质具有决定性作用,其分别为乳杆菌属(51.06%)、乳球菌属(30.29%)、醋酸菌属(7.13%)和巨型球菌属(0.70%)。
3 结 论
通过454焦磷酸高通量测序技术,得到了足够的细菌16S rRNA基因高质量序列,经分析揭示了酸马奶传统发酵过程中细菌多样性变化情况。酸马奶发酵过程中存在丰富的细菌多样性,其中硬壁菌门和变形菌门为优势细菌门,乳杆菌属为优势细菌属。
在酸马奶发酵过程中,细菌群落结构存在显著的演替变化。发酵初期细菌多样性最高,主要来源于发酵剂。在发酵过程中不同菌群之间存在激烈的生存竞争和互生关系。酸马奶发酵微生物中酵母菌[24]也是重要组成部分,将酵母菌等真核微生物一同进行分析是今后研究的重点。
基于非培养方法的分子生物学手段研究细菌多样性虽然有着诸多优点,但是基因测序只能知道相对物种丰度,只能得到数据库里已知物种无法确定新物种,无法得到菌体。因此在研究中应结合分离培养、形态观察、生理生化特征鉴定等传统方法,互相取长补短,才能更深入地揭示酸马奶中微生物世界的奥秘。
[1] WOOD B J B. Microbiology of fermented foods[M]. 2nd ed. London: Elsevier Applied Science Publishers, 1985: 308-345.
[2] 孙天松, 王俊国, 张列兵. 中国新疆地区酸马奶中乳酸菌生物多样性研究[J]. 微生物学通报, 2007, 34(3): 451-454. DOI:10.3969/ j.issn.0253- 2654.2007.03.013.
[3] 乌日娜. 内蒙古传统酸马奶中乳杆菌的分离鉴定及 16S rDNA 序列同源性分析[D]. 呼和浩特: 内蒙古农业大学, 2005.
[4] 孟和毕力格, 乌日娜, 王立平. 不同地区酸马奶中乳杆菌的分离及其生物学特性的研究[J]. 中国乳品工业, 2004, 32(11): 6-11. DOI:10.3969/j.issn.1001-2230.2004.11.002.
[5] HAO Y, ZHAO L, ZHANG H, et al. Identification of the bacterial biodiversity in koumiss by denaturing gradient gel electrophoresis and species-specific polymerase chain reaction[J]. Journal of Dairy Science, 2010, 93(5): 1926-1933. DOI:10.3168/jds.2009-2822.
[6] 刘芳, 都立辉, 杜鹏. 内蒙古酸马奶中乳酸菌多样性的研究[J]. 食品科学, 2008, 29(2): 218-224. DOI:10.3321/j.issn:1002-6630.2008. 02.043.
[7] 殷文政, 乌尼, 钱建伟. 锡林郭勒牧区马奶酒生物活性动态的研究[J].内蒙古农业大学学报, 2002, 23(1): 9-16. DOI:10.3969/ j.issn.1009-3575.2002.01.002.
[8] KAMAGATA Y, TAMAKI H. Cultivation of uncultured fastidious microbes[J]. Microbes Enviroments, 2005, 20(2): 85-91. DOI:10.126 4/jsme2.20.85.
[9] CHAKRAVORTY S, HELB D, BURDAY M, et al. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria[J]. Journal of Microbiological Methods, 2007, 69(2): 330-339. DOI:10.1016/j.mimet.2007.02.005.
[10] ZHOU Xiaoguang, REN Lufeng, LI Yuntao, et al. The next-generation sequencing technology: a technology review and future perspective[J]. Science China Life Sciences, 2010, 53(1): 44-57. DOI:10.1007/ s11427-010-0023-6.
[11] ALEGRIA A, SZCZESNY P, MAYO B, et al. Biodiversity in oscypek, a traditional polish cheese, determined by culture-dependent and independent approaches[J]. Applied and Environmental Microbiology, 2012, 78(6): 1890-1898. DOI:10.1128/AEM.06081-11.
[12] LEE O O, WANG Y, YANG J, et al. Pyrosequencing reveals highly diverse and species specifi c microbial communities in sponges from the Red Sea[J]. International Society for Microbial Ecology, 2011, 5(4): 650-664. DOI:10.1038/ismej.2010.165.
[13] KUCZYNSKI J, STOMBAUGH J, WALTERS W A, et al. Using QIIME to analyze 16S rRNA gene sequences from microbial communities[J]. Current Protocols in Microbiology, 2012, 11(1): 1-20. DOI:10.1002/9780471729259.mc01e05s27.
[14] CAPORASO J G, BITTINGER K, BUSHMAN F D, et al. PyNAST: a flexible tool for aligning sequences to a template alignment[J]. Bioinformatics, 2010, 26(2): 266-267. DOI:10.1093/bioinformatics/ btp636.
[15] HAAS B J, GEVERS D, EARL A M, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J]. Genome Research, 2011, 21(3): 494-504. DOI:10.1101/gr.112730.110.
[16] COLE J R, CHAI B, FARRIS R J, et al. The ribosomal database project (RDP-II): introducing my RDP space and quality controlled public data[J]. Nucleic Acids Research, 2007, 35: 169-172. DOI:10.1093/nar/gkl889.
[17] PRICE M N, DEHAL P S, ARKIN A P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix[J]. Molecular Biology and Evolution, 2009, 26(7): 1641-1650. DOI:10.1093/molbev/msp077.
[18] LOZUPONE C, KNIGHT R. UniFrac: a new phylogenetic method for comparing microbial communities[J]. Applied and Environmental Microbiology, 2005, 71(12): 8228-8235. DOI:10.1128/ AEM.71.12.8228-8235.2005.
[19] 海娜. 白酒发酵过程中原核微生物群落分析[D]. 呼和浩特: 内蒙古大学, 2013.
[20] SANG H J, JI Y J, SE H L, et al. Microbial succession and metabolite changes during fermentation of dongchimi, traditional Korean watery kimchi[J]. International Journal of Food Microbiology, 2013, 164(1): 46-53. DOI:10.1016/j.ijfoodmicro.2013.03.016.
[21] XU Haiyan, SUN Zhihong, LIU Wenjun, et al. Assessment of the bacterial and fungal diversity in home-made yoghurts of Xinjiang, China by pyrosequencing[J]. Journal of the Science of Food & Agriculture, 2015, 95(10): 2007-2015. DOI:10.1002/jsfa.6912.
[22] 孙健. 酸马奶酒中抗菌物质的分离及抗菌作用的研究[D]. 呼和浩特: 内蒙古农业大学, 2003.
[23] 唐传核, 彭志英. 浅析大豆发酵食品的功能性成分[J]. 中国酿造, 2000, 19(5): 8-10. DOI:10.3969/j.issn.0254-5071.2000.05.004.
[24] 倪慧娟, 包秋华, 孙天松. 新疆地区酸马奶中酵母菌多样性研究[J].微生物学报, 2007, 47(4): 578-582. DOI:10.3321/j.issn:0001-6209. 2007.04.003.
Dynamic Changes of Bacteria Community Structure during Koumiss Fermentation
Burenqiqige1,2, GAO Yahan1, REN Xiujuan1, BAO Yanqing1, WEI Ruiyuan1, ZHAO Yiping1, HAN Haige1, Wuyundalai1, HUANG Jinlong1, Dugarjaviin Manglai1,*
(1. Inner Mongolia Mongolian Horse Genetic Resources Protection and Industrial Engineering Laboratory, College of Animal Science, Inner Mongolia Agricultural University, Hohhot 010018, China; 2. Department of Basic Medical, Inner Mongolia Medical University, Hohhot 010110, China)
Samples of koumiss during the fermentation period were taken for the extraction of metagenomic DNA. PCR amplifi cation of the 16S rRNA gene sequence was conducted followed by 454 pyrosequencing to analyze the succession of bacterial community structure. The main fi ndings were as follows: 1) The highest diversity of bacteria occurred at the early stage of fermentation, while bacterial abundance showed the maximum value at 72 h. 2) Firmicutes and Proteobacteria were the dominant phyla in koumiss fermentation. Lactobacillus and Lactococcus were the dominant bacterial genera. 3) With the extension of fermentation time, there were rising or falling trends in bacterial counts at phylum and genus levels. This study has provided a new understanding of dynamic changes in bacterial community structure during koumiss fermentation, which may offer some
for exploring bacterial community structure in other fermented dairy products.
koumiss; traditional fermentation; bacterial community structure; 16S rRNA
10.7506/spkx1002-6630-201611019
TS252.56
A
2015-11-24
国家科学技术部对俄科技合作专项(2011DFR30860);内蒙古自治区科技厅重点实验室建设项目(20130902);内蒙古自治区科技厅应用技术研究与开发项目(20140172);呼和浩特市科技计划项目(2014-农-16)
布仁其其格(1982—),女,讲师,博士研究生,研究方向为分子数量遗传学。E-mail:qiqigejin@163.com
*通信作者:芒来(1962—),男,教授,博士,研究方向为分子数量遗传学与马的育种。E-mail:dmanglai@163.com
