1-乙胺基-3-甲基咪唑四氟硼酸盐吸收CO2的理论研究
2016-11-03张红梅王连军沈锦优
张 慧, 张红梅, 王连军, 沈锦优
(1. 南京理工大学环境与生物工程学院, 南京 210094;2. 南京信息工程大学江苏省大气环境与装备技术协同创新中心, 南京 210044)
1-乙胺基-3-甲基咪唑四氟硼酸盐吸收CO2的理论研究
张慧1,2, 张红梅1, 王连军1, 沈锦优1
(1. 南京理工大学环境与生物工程学院, 南京 210094;2. 南京信息工程大学江苏省大气环境与装备技术协同创新中心, 南京 210044)
采用密度泛函理论(DFT)对离子液体1-乙胺基-3-甲基咪唑四氟硼酸盐([NH2e-mim][BF4])吸收CO2的反应机理进行了研究. 在B3LYP/6-311++G(d,p)计算水平下, 对离子液体[NH2e-mim][BF4]的结构及与CO2反应的中间体、 过渡态和产物进行了全优化, 获得了优化结构的构型参数、 振动频率和热力学数据. 利用自然键轨道(NBO)分析了离子液体[NH2e-mim][BF4]和CO2的自然电荷布居. 计算结果表明, 通过阳离子[NH2e-mim]+自偶解离产生的阳离子[NH3e-mim]2+能与阴离子[BF4]-结合形成更强的离子键. 根据反应吉布斯自由能变(ΔG0—)和焓变(ΔH0—)的计算结果, 判断离子液体[NH2e-mim][BF4]吸收CO2按理论摩尔比2∶1分步进行反应, 吸收过程中质子的转移需克服52.51 kJ/mol的能垒.
离子液体; 二氧化碳; 密度泛函理论; 吸收机理
离子液体具有良好的化学稳定性和热稳定性、 液程宽、 蒸汽压低及不易挥发等特点, 被认为是传统有机试剂的理想替代物, 而且离子液体的结构具有可设计性, 即根据一定的目的设计合成含有特定官能团的功能型离子液体. 如含有碱性基团的功能型离子液体在常温常压下对CO2等酸性气体的溶解能力强, 且具有产物易于分离及循环使用性高等特性, 比传统的醇胺类有机吸收剂更有优势, 被认为在温室气体CO2回收利用方面具有广泛的应用前景. 但目前功能型离子液体合成复杂, 纯化困难, 产品的纯度与产率均较低, 无法对有限合成的功能型离子液体的结构、 性质及反应机理进行深入研究, 使此类离子液体的进一步推广和应用受到了极大的限制.
近几十年来, 量子化学计算已被广泛应用于物质的分子结构、 物化性质的预测及化学反应机理等方面的研究[1~4]. 目前, 量子化学计算方法主要有半经验(Semi-empirical)法、 从头计算(Abinitio)法、 基于微扰理论的MP方法及密度泛函理论(DFT)方法. 其中, DFT是最常用的量子化学计算方法, 它已成功应用于有机大分子[5~7]和固体功能材料[8,9]的结构和热力学性质以及微观反应机理的研究[10]. 在功能型离子液体的理论研究方面, Li等[11]采用DFT方法对羟烷基胺离子液体(HyAAILs)与SO2的相互作用和吸收机理进行了研究, 结合实验确定了该离子液体吸收SO2的理论摩尔分数. Zhang等[12]采用DFT方法在B3LYP/6-311++G**计算水平下对1-丙基-3-甲基咪唑谷氨酸液体([C3mim][Glu])的阴阳离子间相互耦合作用及相关性质进行了研究. Sun等[13]采用DFT方法对1,2-二甲基-3-胺乙基咪唑四氟硼酸盐[aEMMIM][BF4]捕集CO2的机理进行了研究. 以上研究结果表明, DFT方法在化学领域尤其对离子液体的研究是一种有效且重要的方法, 通过量子化学计算研究离子液体将有助于加深在微观层次上对离子液体分子结构特征及其化学反应机理的认识与理解.
本文采用DFT方法对1-乙胺基-3-甲基咪唑四氟硼酸盐离子液体([NH2e-mim][BF4])的结构及其吸收CO2的可能反应过程进行了理论计算, 以探究离子液体的自偶解离和[NH2e-mim][BF4] 吸收CO2的反应机理, 进而从理论上为功能型离子液体的分子设计和吸收气体的机理研究提供参考.
1 计算方法
通常, DFT中的B3LYP方法不适于研究范德华力为主导的弱相互作用体系, 这是由于该方法虽然考虑了电子的相关性, 但是它忽略了相互作用体系的色散力, 而色散作用在弱相互作用体系中显得尤为重要, 是不能被忽略的, 否则相互作用能的计算可能出现较大误差[13~15]. 但对于离子液体, 已有研究[16,17]结果表明离子液体中阴阳离子之间的相互作用能远远大于一般弱相互作用体系. 因此本文忽略了色散作用对离子液体-CO2体系的反应Gibbs自由能和焓变的贡献, 而是在单点能计算的基础上, 对体系的总能量进行了零点能(ZPE)校正. 孙小丽等[18]从氢气吸附的实验对密度泛函的基准进行了研究, 发现B3LYP的计算误差最小. Song等[19]和Wang等[20]的研究也表明B3LYP泛函具有很小的计算误差. 这些研究表明采用DFT中的B3LYP方法研究离子液体-CO2体系是可以满足要求的. 在基函数的选择方面, B3LYP泛函常用的基函数包括6-31G(d)、 6-31G(d,p)、 6-311++G(d,p)和6-311++G(3df,2pd)等, 通常所选基函数越大, 计算精度越高, 但是计算耗时却越长. 为了获得较为精确的计算结果, 同时考虑适用性和实用性, 通常在功能离子液体理论研究方面倾向于选择6-311++G(d,p)基组[12,13,18,21,22], 以上研究结果表明该基组计算精度和计算结果的可靠程度都较高.
鉴于此, 本文选择了在DFT/B3LYP/6-311++G(d,p)计算条件下对离子液体[NH2e-mim]BF4与CO2反应体系中可能涉及到的反应物、 中间产物、 过渡态、 产物的结构、 频率及热力学性质进行了理论计算, 获得各物质优化后的稳定结构、 能量、 频率及自然电荷布居(NBO电荷)等.
文中所有的计算均采用Gaussian 09程序包[23]完成. 文中所获能量均通过零点能进行校正, 反应中除过渡态外, 所有物质都被确认为只有实频, 而过渡态则有且只有一个虚频. 另外, 反应中的过渡态采用OPT=TS方法不断优化而得到, NBO电荷布居分析采用Gaussian 09自带的NBO程序完成. 文中涉及反应的吉布斯自由能变(ΔG0—)和焓变(ΔH0—)均由化学中经典的热力学公式求得.
2 结果与讨论
2.1结构优化
在DFT/B3LYP/6-311++G(d,p)计算水平下对离子对[NH2e-mim][BF4]、 阳离子[NH2e-mim]+、 阴离子[BF4]-及CO2的初始结构进行了全优化, 优化后的稳定构型如图1所示. 对应构型的部分结构参数(键长和键角)列于表1.
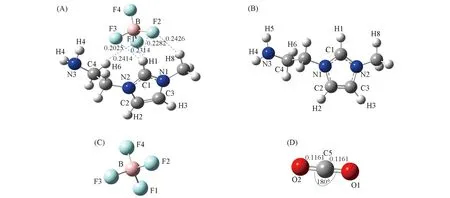
Fig.1 Optimized geometric configurations and some geometrical parameters of [NH2e-mim][BF4](A), [NH2e-mim]+(B), [BF4]-(C) and CO2(D) Bond length or distance/nm, angle/(°).
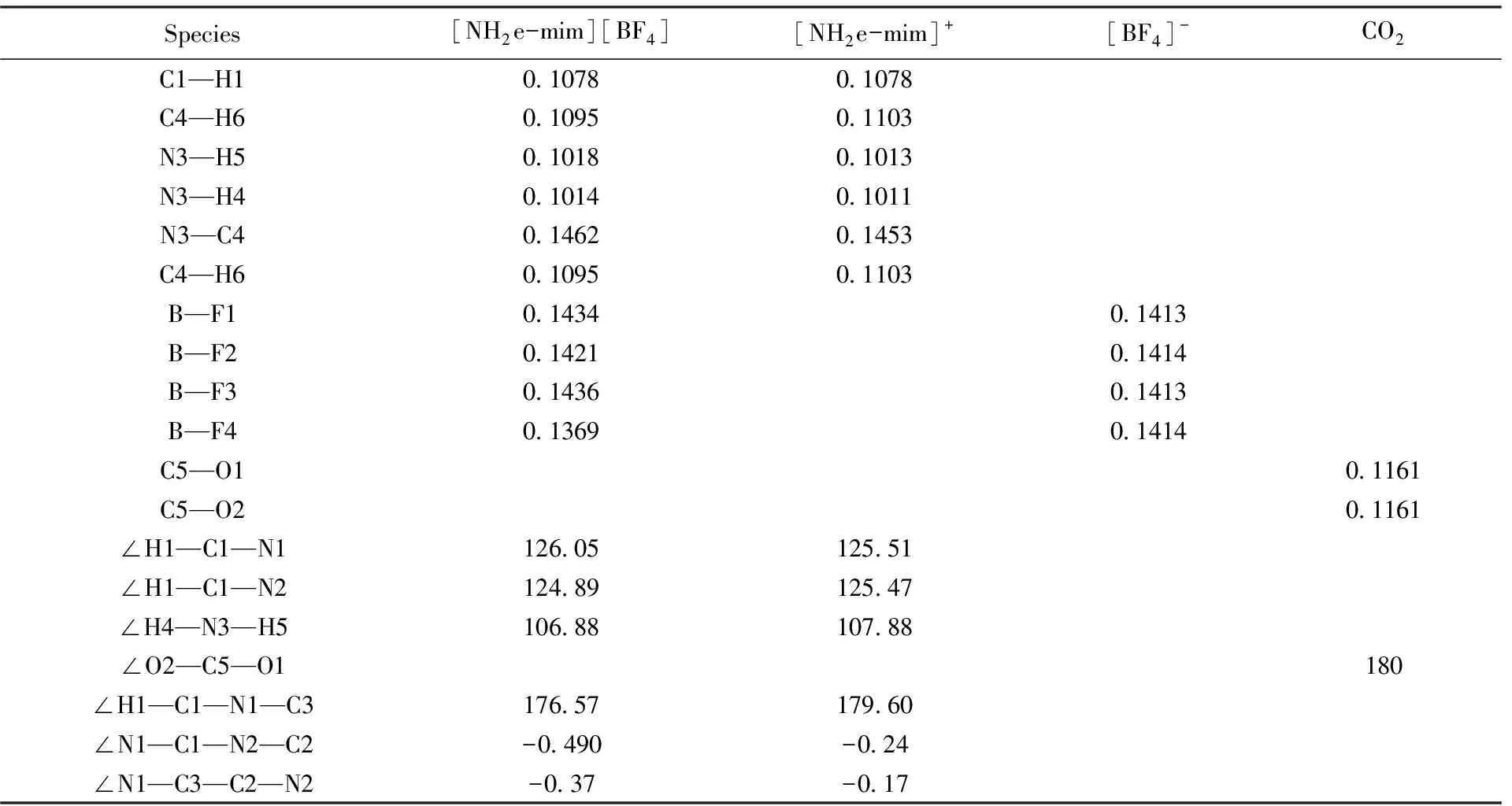
Species[NH2e-mim][BF4][NH2e-mim]+[BF4]-CO2C1—H10107801078C4—H60109501103N3—H50101801013N3—H40101401011N3—C40146201453C4—H60109501103B—F10143401413B—F20142101414B—F30143601413B—F40136901414C5—O101161C5—O201161∠H1—C1—N11260512551∠H1—C1—N21248912547∠H4—N3—H51068810788∠O2—C5—O1180∠H1—C1—N1—C31765717960∠N1—C1—N2—C2-0490-024∠N1—C3—C2—N2-037-017


由于离子液体阴阳离子间主要是离子键作用, 因此当二者之间距离较远时, [NH2e-mim]+[BF4]-有电离反应:
(1)
解离产生的阳离子[NH2e-mim]+因含有既是质子酸又是质子碱的氨基, 因此它还可能进一步发生质子自递反应:
(2)
本文对离子液体[NH2e-mim][BF4]-CO2反应体系中的主要离子及离子对结构在298.15 K, 1.0×105Pa条件下的热力学性质进行了考察, 获得了各优化结构的热力学数据及零点能(ZPE), 结果列于表2. 以表2数据为基础, 再计算各反应式的ΔG0—和ΔH0—(见表3). 由表3可见, 在电离过程中反应式(1)和(2)的ΔG0—分别为332.65和553.98 kJ/mol, ΔH0—分别为385.16和701.01 kJ/mol, 说明[NH2e-mim][BF4]的电离及[NH2e-mim]+的自耦电离反应均为吸热过程.
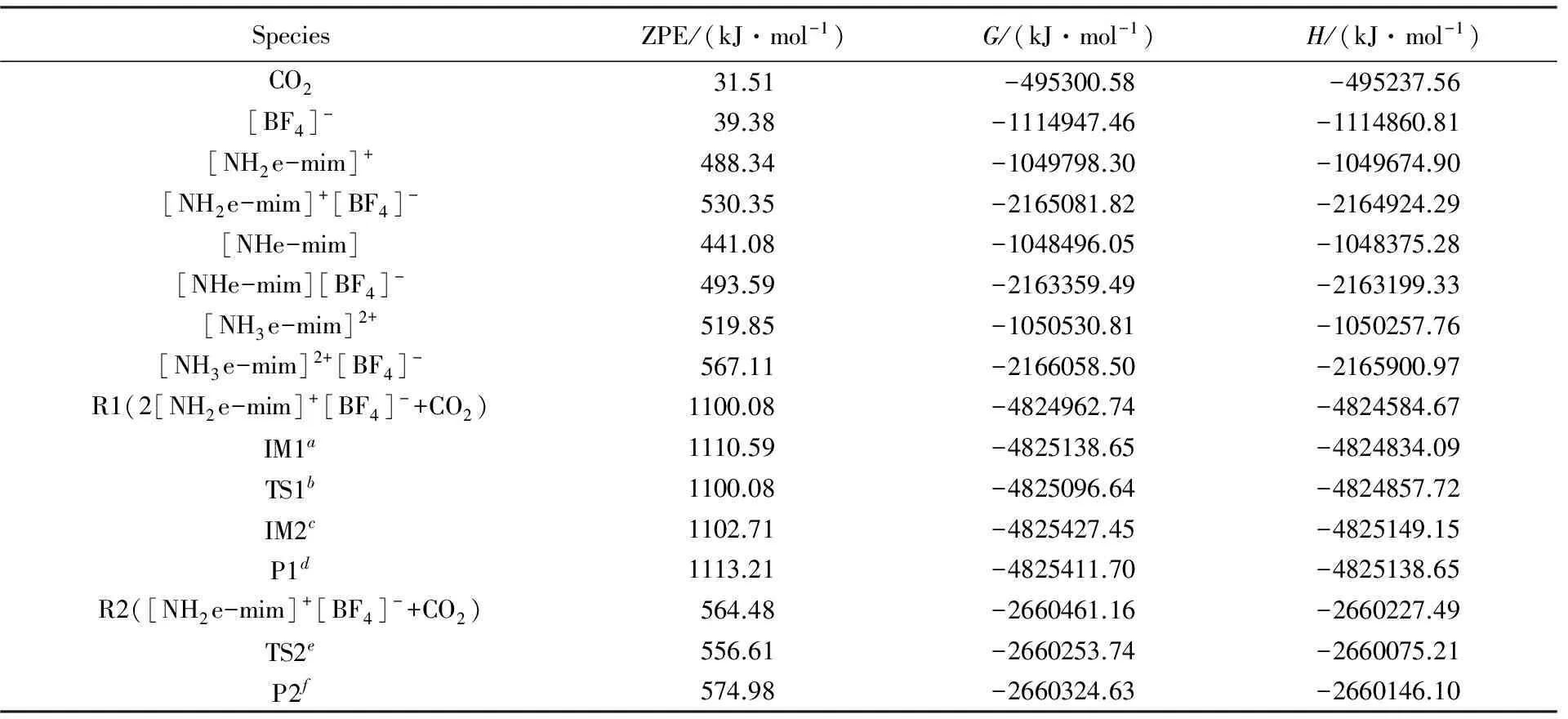
Table 2 Thermodynamic parameters of optimized configurations in [NH2e-mim]BF4-CO2reaction system
a. The first intermediate(IM1) of [NH2e-mim]+[BF4]-absorbing CO2with the theoretical molar ratio of 2∶1, denoted as [mim-eNH2COO…NH2e-mim]2+2[BF4]-;b. the first transition state(TS1) of [NH2e-mim]+[BF4]-absorbing CO2with the theoretical molar ratio of 2∶1, denoted as [mim-eNHCOO-…H…NH2e-mim]2+2[BF4]-;c. the second intermediate(IM2) of [NH2e-mim]+[BF4]-absorbing CO2with the theoretical molar ratio of 2∶1, denoted as [mim-eNHCOO—H…NH2+e-mim]2+2[BF4]-;d. the product(P1 ) of [NH2e-mim]+[BF4]-absorbing CO2with the theoretical molar ratio of 2∶1, denoted as [mim-eNHCOO—NH3+e-mim]2+2[BF4]-;e. the first transtion state(TS2) of [NH2e-mim]+[BF4]-absorbing CO2with the theoretical molar ratio of 1∶1, denoted as [COOH…NHe-mim]+[BF4]-;f. the product(P2) of [NH2e-mim]+[BF4]-absorbing CO2with the theoretical molar ratio of 1∶1, denoted as [HOOC—NHe-mim]+[BF4]-.

Table 3 ΔG 0— and ΔH 0— for each reaction
2.2离子键比较
在[NH2e-mim][BF4]-CO2反应体系中, 阴离子[BF4]-与[NHe-mim], [NH2e-mim]+和[NH3e-mim]2+之间也可能发生如下的结合反应:
(3)
(4)
(5)
由表3可见, 在各离子结构的结合过程中, 反应(3)~(5)的ΔG0—分别为97.93, -332.65和-571.57 kJ/mol, ΔH0—分别为49.88、 -385.16和-773.73 kJ/mol. 对比分析可知, 因[NH3e-mim]2+[BF4]-的离子键比[NH2e-mim]+[BF4]-强, 反应(5)较反应(4)更易发生, 而总电荷为零的[NHe-mim]与[BF4]-的结合较为困难, 因此反应(3)较难自发进行.
2.3离子液体[NH2e-mim][BF4]吸收CO2的反应机理
由于离子液体[NH2e-mim][BF4]的阳离子上有—NH2基团, 因而可能通过化学反应对CO2进行吸收. 在含氨基的功能型离子液体对CO2吸收机理的研究中, 目前认为主要存在两种机制:(1) 离子液体上的—NH2与CO2以摩尔比2∶1反应, 生成氨基甲酸盐, 如氨基咪唑类离子液体[NH2p-bmim]·[BF4]与CO2的反应过程[27~29]; 或如季胺类氨基酸盐离子液体[N4444][β-Ala]与CO2的反应过程[30]; (2) 离子液体上的—NH2与CO2以摩尔比1∶1反应, 生成氨基甲酸, 如季膦类氨基酸盐离子液体[P66614][Met]吸收CO2的反应[31]. 不同的吸收机理将导致功能型离子液体CO2吸收量的差异. 本文尝试从理论上探究离子液体[NH2p-bmim][BF4]对CO2的2种可能吸收机理2∶1(R1)和1∶1(R2), 反应表达式如图2所示. 表3计算结果显示, [NH2e-mim][BF4]与CO2以摩尔比1∶1反应, 其ΔG0—和ΔH0—分别为136.53 和81.39 kJ/mol, 表明该反应在常温常压下很难自发进行, 且为吸热反应; 而[NH2e-mim][BF4]与CO2以摩尔比2∶1反应, 其ΔG0—和ΔH0—分别为-448.96和-553.19 kJ/mol, 表明该反应不仅能在常温常压下自发进行, 且为放热反应; 同时, 2种反应的势能图(图4)也表明反应途径R2的能量比反应途径R1的低, 这也说明了从化学热力学的角度判断离子液体以摩尔比2∶1吸收CO2更易进行, 这一结论与文献[32~34]的结果一致. 因此, 本文重点讨论了[NH2e-mim][BF4]吸收CO2的2∶1(R1)反应过程.

Fig.2 Possible reaction mechanism of functional ionic liquid containing —NH2 capture CO2(A) Pathway R1; (B) pathway R2.
2.3.1反应过程中几何构型及能量的变化图3为经过多次优化后得到的[NH2e-mim]BF4-CO2反应过程中各反应物、 中间体、 过渡态及产物的几何构型, 同时标示了各物质中一些重要部位的构型参数, 以便对比这些构型参数在反应过程中的变化. 图4反映了2种吸收过程中体系能垒的变化情况, 以此为基础可以对反应过程中相关物质的相对能量变化进行描述. 值得注意的是, 一般情况下DFT方法会低估反应的能垒等计算数值, 但这对于判断体系整体能量变化的趋势影响并不大.
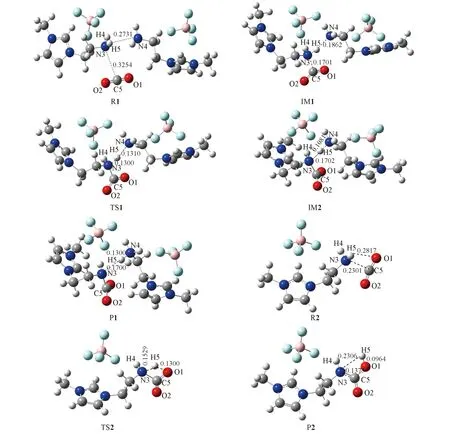
Fig.3 Geometric configuration changes in [NH2e-mim][BF4]-CO2 reaction system

Fig.4 Energy barrier(kJ/mol) variation of [NH2e-mim]BF4-CO2 reaction process

Fig.5 Natural charge(NBO) distribution of [NH2e-mim][BF4](A) and CO2(B)
[NH2e-mim][BF4]的N3与CO2分子的C5原子成键后, 暂时形成中间体IM1中一个氮原子上带有2个氢原子的氨基甲酸根. 从图3的TS1构型可见, 与N3相连的H5在N3与另一个阳离子[NH2e-mim]的N4之间来回迁移. 其中, N3—H5的键长由原来的0.1080 nm增大至0.1300 nm, 而H5与N4的距离也由中间体IM1中的0.1862 nm缩短为0.1310 nm. 表明N3—H5即将断开, H5—N4将要形成. 图4显示过渡态的能量高出反应物52.51 kJ/mol, 此时反应体系的能量达到最高, 体系状态最不稳定. 通常, 由中间体到过渡态的过程对于反应的进行极为重要, 该阶段的能垒如果过高将导致反应无法进行. 计算结果显示, [NH2e-mim][BF4]-CO2的反应过程中IM1到TS1的反应能垒约为36.76 kJ/mol. 明显小于吸收途径R2要跨过的能垒97.94 kJ/mol, 说明从化学热力学角度判断[NH2e-mim][BF4]吸收CO2的反应按途径R1比较容易进行. 途径R1中极为活泼的过渡态很不稳定, 会很快向产物或反应物转变. 当质子H5向另一[NH2e-mim][BF4]氨基的N4原子迁移靠近时, 形成中间体IM2(图3), 该中间体的H5与N4的距离进一步缩短为0.1081 nm, 是N—H键的成键距离, 与产物的N4—H5键的键长基本相同. 此时中间体IM2的能量处于过程最低值, 但仍不稳定, 将进一步转变以形成稳定的产物P1.
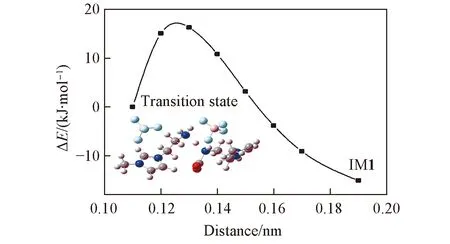
Fig.6 Relaxation scanning energy curve of N3—H5 in IM1
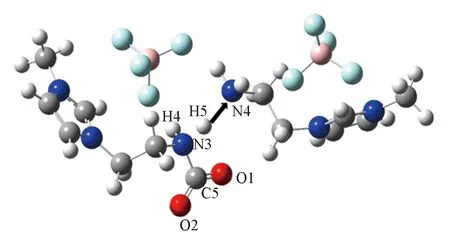
Fig.7 Proton(H5) transfer from N3 to N4 in [NH2e-mim]BF4-CO2 reaction system
2.3.2质子转移过程分析鉴于形成初始复合物是离子液体捕获CO2的第一步, 本文重点对反应中的质子转移过程进行了考察. 过渡态R1-TS1有且仅有一个虚频(992.97i cm-1), 由对中间体IM1沿着N3—H5键进行松弛扫描得到的能量E与原子间距dN3—H5的关系曲线(图6)可知, CO2的C5与[NH2e-mim]BF4的N3接近成键的过程中(dN3—H5=0.110~0.190 nm)存在过渡态的一阶鞍点, 从而证实了该过渡态结构的真实性. 图6给出了解析过程中反应体系的总能量随N3—H5间距的变化情况. 由图6可见, 随着反应的进行, N3—H5间的距离不断地增大, 而N4—H5间的距离逐渐缩短, 此时CO2上的C5已与N3成键. 表明离子液体[NH2e-mim][BF4]在吸收CO2的过程中, 首先是CO2上的C原子攻击[NH2e-mim][BF4]中乙胺基的N, 然后该基团的一个质子H转移到另一个[NH2e-mim][BF4]中乙胺基的N上, 该过程的分步机理如图7所示.
3 结 论
采用量子化学计算对[NH2e-mim][BF4]与CO2反应的反应物、 中间体、 过渡态和产物的结构在DFT/B3LYP/6-311++G(d,p)基组水平下进行结构的全面优化、 能量及频率的计算. 计算结果表明, 阳离子[NH2e-mim]+自偶解离产生的阳离子[NH3e-mim]2+比[NH2e-mim]+更易与阴离子[BF4]-结合, 生成更稳定的离子化合物[NH3e-mim]2+[BF4]-. 根据离子液体吸收CO2反应过程可能的能量变化, 确定吸收反应机理:即从热力学的角度, 离子液体[NH2e-mim][BF4]吸收CO2的反应更易以摩尔比2∶1的分步反应进行, 过程是CO2中的C首先进攻离子液体中乙胺基上的N原子, 形成一个C—N键后, 该—NH2上的一个H质子转移到另一个[NH2e-mim][BF4]的—NH2上; 该质子转移过程克服52.51 kJ/mol的能垒后形成了一个[O2C—NHe-mim]氨基甲酸两性离子和一个[NH3e-mim]2+阳离子, 最终形成产物[mim-eNHCOO-—NH3+e-mim]2+2[BF4]-加合物.
[1]Ahmed D., Ludwik A., Guido M.,J.Phys.Chem.A, 2000, 104, 2112—2119
[2]Vincenzo B.,J.Phys.Chem.A, 2004, 108, 4146—4150
[3]Schlu1cker S., Singh R. K., Asthana B. P., Popp J. Kiefer W.,J.Phys.Chem.A, 2001, 105, 9983—9989
[4]Wu Y., Xue Y., Xie D. Q., Yan G. S.,J.Org.Chem., 2005, 70, 5045—5054
[5]Liu T. T., Lu X., Zhang M. T.,Chem.Res.ChineseUniversities, 2014, 30(4), 656—660
[6]Wang P. C., Lu M.,Chem.J.ChineseUniversities, 2014, 35(3), 596—601(王鹏程, 陆明. 高等学校化学学报, 2014, 35(3), 596—601)
[7]Li Y., Jian L.,Chem.Res.ChineseUniversities, 2014, 30(6), 997—1004
[8]Yu Y. X.,Phys.Chem.Chem.Phys., 2013, 15, 16819—16827
[9]Wu D. L., Jiang W., Liu X. Q., Qiu N. X., Xue Y.,Chem.Res.ChineseUniversities, 2016, 32(1), 118—126
[10]Gholizadeh R., Yu Y. X.,Appl.Surf.Sci., 2015, 357, 1187—1195
[11]Li X. L., Chen J. J., Luo M., Chen X. Y., Li P. P.,ActaPhys. -Chim.Sin., 2010, 26(5), 1364—1372
[12]Zhang Y., Chen X. Y., Wang H. J., Diao K. S., Chen J. M.,J.Mol.Struc-Theochem., 2010, 952, 16—24
[13]Sun H., Zhou X.Q., Xue Z. M., Zhou Z. Y., Mu T. C.,Int.J.Greenh.GasCon., 2014, 20, 43—48
[14]Yu Y. X.,ACSAppl.Mater.Interfaces, 2014, 6, 16267—16275
[15]Yu Y. X.,J.Mater.Chem.A, 2014, 2, 8910—8917
[16]Magalhaes A. L., Madail S. R., Ramos M. J.,Theor.Chem.Acc., 2000, 105, 68—76
[17]Loos P. F., Assfeld X., Rivail J. L.,Theor.Chem.Acc., 2007, 118, 165—171
[18]Sun X. L., Huo R. P., Bu X. Y., Li J. L.,Chem.J.ChineseUniversities, 2015, 36(8), 1570—1575(孙小丽, 霍瑞萍, 步宇翔, 李吉来. 高等学校化学学报, 2015, 36(8), 1570—1575)
[19]Song Z. X., Wang H. J., Xing L. J.,J.Struct.Chem., 2009, 20, 509—515
[20]Wang C. M., Zheng J. J., Cui G. K., Luo X. Y., Guo Y., Li H. R.,Chem.Commun., 2013, 49, 1166—1168
[21]Gao H. Y., Zhang Y., Wang H. J., Liu J. H., Chen J. M.,J.Phys.Chem., 2010, 114, 10243—10252
[22]Christopher A. Z., Gary S. G., Michael T. B., James D., Tetsuya T., Rika H.,J.Am.Soc.MassSpectrom., 2015, 26, 1559—1569
[23]Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J., R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam N. J., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E. Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J.,Gaussian09,RevisionA. 1, Gaussian Inc., Wallingford CT, 2009
[24]Herzberg G.,ElectronicSpectraofPolyatomicMolecules, Van Nostrand, New York, 1996
[25]Babarao R., Dai S.,J.Phys.Chem.B, 2011, 115(32), 9789—9794
[26]Liu K. H., Pu M., Li H. Y., Chen B. H.,Chin.J.Chem.Phys., 2005, 18(3), 332—335(刘坤辉, 蒲敏, 李会英, 陈标华. 化学物理学报, 2005, 18(3), 332—335 )
[27]Yu G. R., Zhang S. J., Yao X. Q., Zhang J. M., Dong K., Dai W. B., Mori R. H.,Ind.Eng.Chem.Res., 2006, 45(8), 2875—2880
[28]Katsyuba S. A., Griaznova T. P., Vidiš A., Dyson P. J.,J.Phys.Chem.B, 2009, 113, 5046—5051
[29]Bates E. D., Mayton R. D., Ntai I., Davis J. H.,J.Am.Chem.Soc., 2002, 124(6), 926—927
[30]Zhang J. M., Zhang S. J., Dong K., Zhang Y. Q, Shen Y. Q., Lv X. M.,Chem.Eur.J., 2006, 12(15), 4021—4026
[31]Gurkan B. E., de la Fuente J. C., Mindrup E. M., Ficke L. E., Goodrich B. F., Price E. A., Schneider W. F., Brennecke J. F.,J.Am.Chem.Soc., 2010, 132(7), 2116—2117
[32]Zhang Y. Q., Zhang S. J., Lu X. M., Zhou Q., Fan W., Zhang X. P.,Chem.Eur.J., 2009, 15, 3003—3011
[33]Zhang J. M., Zhang S. J., Dong K., Zhang Y. Q., Shen Y. Q., Lv X. M.,Chem.Eur.J., 2006, 15, 4021—4026
[34]Li X. Y., Hou M. Q., Zhang Z. F., Han B. X., Yang G. Y., Wang X. L., Zou L. Z.,GreenChem., 2008, 10, 879—884
(Ed.:Y, Z, S)
† Supported by the Natural Science Research Project of Colleges and Universities in Jiangsu Province, China(No.12KJB610003).
Density Functional Theory Studies on the CO2Absorption by 1-Ethylamine-3-methylimidazolium Tetrafluoroborate†
ZHANG Hui1,2*, ZHANG Hongmei1, WANG Lianjun1, SHEN Jinyou1
(1. School of Environmental and Biological Engineering, Nanjing University ofScienceandTechnology,Nanjing210094,China;2.JiangsuCollaborativeInnovationCenterofAtmosphericEnvironmentandEquipmentTechnology(CICAEET),NanjingUniversityofInformationScience&Technology,Nanjing210044,China)
CO2absorption mechanism by ionic liquids 1-ethylamine-3-methylimidazolium tetrafluoroborate, which was formulated as [NH2e-mim][BF4], was describedviadensity functional theory(DFT). The structure of ionic liquids [NH2e-mim][BF4], their reaction intermediates, transition states and products, were optimized using the B3LYP/6-311++G(d,p) basis method, with the optimized configuration parameters, vibration frequencies and thermodynamics data obtained. Furthermore, the natural bond orbital atomic charge assignments were also calculatedviathe natural bond orbital(NBO) method. The computational results demonstrated that the divalent cation [NH3e-mim]2+, which was produced by the autoprotolysis of cation [NH2e-mim]+, could be easily combined with anion [BF4]-, with stronger ionic bond formed. According to the calculation results of standard Gibbs free energy(ΔG0—) and enthalpy(ΔH0—), it could be inferred that the absorption of CO2onto ionic liquids [NH2e-mim][BF4] was step by step, in accordance with the theoretical molar ratio of 2∶1. During the absorption process, the energy barrier of 52.51 kJ/mol should be overcome for proton transfer reaction.
Ionic liquid; CO2; Density functional theory(DFT); Absorption mechanism
10.7503/cjcu20160337
2016-05-12. 网络出版日期:2016-08-26.
江苏省高校自然科学研究项目(批准号:12KJB610003)资助.
O641
A
联系人简介:张慧, 女, 讲师, 主要从事大气污染控制新技术及新材料的开发与研究. E-mail:zhanghui13401@163.com
