高效液相色谱法测定腰痛丸(Ⅰ)中新乌头碱、次乌头碱和乌头碱的含量
2016-11-02朱干红仇其原曹桂萍吴士龙
朱干红,仇其原,曹桂萍,吴士龙
(1.盐城市中医院药剂科,江苏 盐城 224001; 2.盐城市药品检验所药物分析室,江苏 盐城 224001)
高效液相色谱法测定腰痛丸(Ⅰ)中新乌头碱、次乌头碱和乌头碱的含量
朱干红1*,仇其原2#,曹桂萍1,吴士龙2
(1.盐城市中医院药剂科,江苏 盐城224001; 2.盐城市药品检验所药物分析室,江苏 盐城224001)
目的:建立腰痛丸(Ⅰ)中新乌头碱、次乌头碱和乌头碱含量测定方法,进一步提高腰痛丸(Ⅰ)的质量控制。方法:采用CAPCELL PAK C18(4.5 mm×250 mm,5 μm)色谱柱,流动相A为乙腈-四氢呋喃(V∶V=25 ∶15),流动相B为0.1 mol/L醋酸铵溶液,梯度洗脱,检测波长为235 nm,柱温为30 ℃,流速为1.0 ml/min。结果:新乌头碱在0.2~1.6 μg/ml范围内线性关系良好,r2=0.999 1,平均回收率为97.69%,RSD为0.86%;次乌头碱在0.2~1.6 μg/ml范围内线性关系良好,r2=0.998 9,平均回收率为98.17%,RSD为0.93%;乌头碱在0.2~1.6 μg/ml范围内线性关系良好,r2=0.999 5,平均回收率为99.50%,RSD为0.45%。结论:本研究测定方法简便、准确、重复性好,可用于腰痛丸(Ⅰ)的质量控制。
高效液相色谱法; 腰痛丸(Ⅰ); 新乌头碱; 次乌头碱; 乌头碱
腰痛丸(Ⅰ)是一种中药药丸,主要是由独活、苍术、干姜、肉桂、陈皮、威灵仙、制川乌、制草乌等中药药材加工提炼而成,具有祛风、散寒、温经、通络、止痛等作用,主治风寒腰痛及关节炎等症。新乌头碱、次乌头碱和乌头碱等乌头类生物碱是存在于川乌、草乌、附子等植物中的主要有毒成分[1],能使神经和心脏中毒。研究发现,口服纯乌头碱0.2 mg即可中毒,3~5 mg可致死。目前对乌头类生物碱的研究多采用高效液相色谱法[2-5]、反相高效液相色谱法[6-7]、液相色谱-串联质谱联用技术[8]、超高效液相色谱-质谱联用技术[9]、液相色谱-质谱联用技术等,其中高效液相色谱法得到的结果更加真实可靠、科学准确[10-11]。乌头类生物碱不稳定,且含量甚微。本研究建立了高效液相色谱法测定腰痛丸(Ⅰ)中新乌头碱、次乌头碱和乌头碱的含量的方法,用于腰痛丸(Ⅰ)的质量控制。
1 材料
1.1仪器与设备
Waters 2695型高效液相色谱仪,配置Waters 2996型二极管阵列紫外检测器、empower处理系统等(美国沃特斯公司);Waters e2695型高效液相色谱仪,配置Waters 2489型紫外检测器等(美国沃特斯公司);减压干燥器(郑州园田数字科技有限公司);AB204-N型电子天平[梅特勒-托利多仪器(上海)有限公司];DGG-9053A型超声波清洗器(常州国华电器有限公司)。
1.2药品与试剂
新乌头碱对照品(批号:110799-200404,含量:100%)、次乌头碱对照品(批号:110798-200805,含量:99.5%,置五氧化二磷减压干燥器中干燥12 h以上使用)、乌头碱对照品(批号:110720-200410,含量:100%)均购自中国食品药品检定研究院;腰痛丸(Ⅰ)(盐城市中医院制剂室,批号:150907、150601、150115,规格:60 g/瓶);异丙醇、三氯甲烷、乙酸乙酯均为分析纯;乙腈、四氢呋喃、醋酸铵、冰醋酸均为色谱纯;水为重蒸馏水。
2 方法与结果
2.1色谱条件与系统适用性
色谱柱:CAPCELL PAK C18(4.5 mm×250 mm,5 μm);流动相:乙腈-四氢呋喃(V∶V=25 ∶15)为流动相A,0.1 mol/L醋酸铵溶液(含冰醋酸0.05%)为流动相B,按表1中的梯度洗脱比例进行梯度洗脱;流速:1.0 ml/min;检测波长:235 nm;柱温:30 ℃;进样量:20 μl;采集时间:70 min。

表1 梯度洗脱比例(%)Tab 1 Ratio of gradient elution(%)
2.2溶液的制备
2.2.1对照品储备液:精密称取新乌头碱对照品9.55 mg、次乌头碱对照品(置于五氧化二磷的减压干燥器中干燥12 h以上)10.50 mg、乌头碱对照品10.45 mg,分别置于10 ml容量瓶中,加入异丙醇-三氯甲烷(V∶V=1 ∶1)混合溶液使其溶解,稀释至刻度,摇匀,即获得新乌头碱对照品储备液、次乌头碱对照品储备液、乌头碱对照品储备液。
2.2.2对照品混合溶液:分别精密量取新乌头碱对照品储备液、次乌头碱对照品储备液和乌头碱对照品储备液各1 ml,置于100 ml容量瓶中,加入异丙醇-三氯甲烷(V∶V=1 ∶1)混合溶液使其溶解,用异丙醇-三氯甲烷(V∶V=1 ∶1)稀释至刻度,摇匀,即获得混合对照品溶液。
2.2.3供试品溶液:称取腰痛丸(Ⅰ)样品30 g,精密称定,于大小适合的研钵中研细,取研细后的细粉10 g,精密称定,置于具塞锥形瓶中,加入氨试液5 ml,精密加入异丙醇-乙酸乙酯(V∶V=1 ∶1)混合溶液100 ml,精密称定质量,超声处理(功率300 W,频率40 kHz,水温在25 ℃以下)30 min,放冷,再精密称定重量,用异丙醇-乙酸乙酯(V∶V=1 ∶1)混合溶液补足减失的重量,摇匀,滤过,残渣用异丙醇-乙酸乙酯(V∶V=1 ∶1)混合溶液50 ml分3次(20、20、10 ml)洗涤,合并滤液于40 ℃以下减压回收溶剂至干,残渣精密加入异丙醇-乙酸乙酯(V∶V=1 ∶1)混合溶液3 ml使其溶解,密塞,滤过,摇匀,取续滤液,即获得供试品溶液。
2.3专属性考察
精密吸取“2.2”项下对照品混合溶液和供试品溶液各20 μl,按“2.1”项下色谱条件注入液相色谱仪,记录色谱图,见图1。由图可见,新乌头碱、次乌头碱和乌头碱三组分色谱峰完全分离,在测定条件下不受其它组分干扰,表明该方法具有良好的专属性。
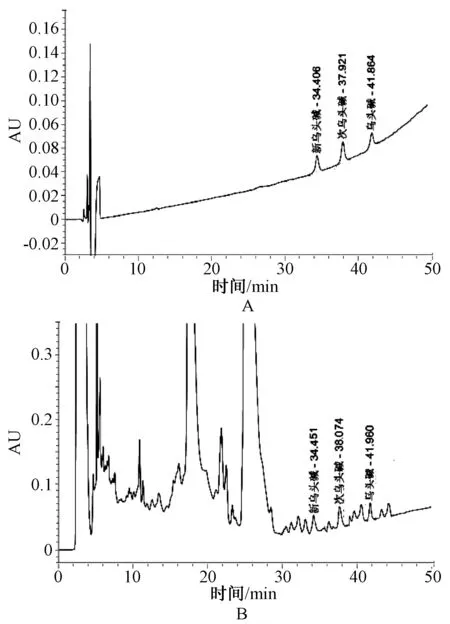
A. 对照品混合溶液;B.供试品溶液A. Control mixed solution; B. Test solution图1 高效液相色谱图Fig 1 HPLC chromatogram
2.4线性关系考察
精密量取“2.2.2”项下对照品混合溶液适量,制成质量浓度分别为0.2、0.4、0.8、1.2、1.6 μg/ml的溶液,按“2.1”项下色谱条件分别进样测定,记录峰面积。以对照品质量浓度(X,μg/ml)对测得的峰面积(Y)进行线性回归,绘制标准曲线。结果表明:新乌头碱在0.2~1.6 μg/ml浓度范围内,线性关系良好,回归方程为:Y=634 897X+2 545.7,r2=0.999 1;次乌头碱在0.2~1.6 μg/ml浓度范围内,线性关系良好,回归方程为:Y=731 225X+1 960.1,r2=0.998 9;乌头碱在0.2~1.6 μg/ml浓度范围内,线性关系良好,回归方程为:Y=488 169X+1 271.9,r2=0.999 5。
2.5精密度试验
分别取“2.2.2”项下对照品混合溶液20.0 μl,按照“2.1”项下色谱条件重复进样6次,记录峰面积,计算RSD。结果,新乌头碱峰面积的RSD=0.94%(n=6),次乌头碱峰面积的RSD=0.86%(n=6),乌头碱峰面积的RSD=0.99%(n=6),表明仪器精密度良好。
2.6稳定性试验
取批号150601的样品适量,按照“2.2.3”项下方法制备供试品溶液,室温(25 ℃)下分别在 0、2、4、8、12、16、18、24 h时按“2.1”项下色谱条件进样测定,记录峰面积,计算RSD。结果,新乌头碱峰面积的RSD=0.06%(n=8),次乌头碱峰面积的RSD=0.08%(n=8),乌头碱峰面积的RSD=0.08%(n=8),表明供试品溶液在24 h内稳定性良好。
2.7重复性试验
取批号150601的样品适量,按照“2.2.3”项下方法制备供试品溶液,共6份,分别精密吸取20 μl,按“2.1”项下色谱条件进样测定,记录峰面积,计算RSD。结果,新乌头碱峰面积的RSD=0.16%(n=6),次乌头碱峰面积的RSD=0.09%(n=6),乌头碱峰面积的RSD=0.21%(n=6),说明本方法重复性良好。
2.8加样回收试验
精密称取已知含量的批号150601样品细粉,共9份,分别精密加入0.8、1.0、1.2 ml标准溶液(标准溶液为新乌头碱、次乌头碱和乌头碱含量为原样品100%含量的溶液),各3份,按照“2.2.3”项下方法制得供试品溶液6 ml,再按“2.1”项下色谱条件进样测定。结果,平均回收率为98.4%,RSD为0.38%,表明加样回收率良好。
2.9供试品含量测定
取各批样品适量,分别按“2.2.3”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定并计算含量,见表2。
表2 样品含量测定结果(n=3,μg/g)Tab 2 Results of content determination of samples(n=3,μg/g)
3 讨论
查阅相关文献发现,乌头碱的含量测定多选择230~254 nm作为检测波长[2-6],其中采用波长235 nm的色谱图基线更加平稳[12-13],检出结果中有干扰作用的的杂质峰少[14-16]。本研究在220~380 nm的波长范围内对新乌头碱、次乌头碱、乌头碱对照品进行光谱扫描,发现3种对照品在235 nm附近有最大吸收波长,故选择235 nm作为检测波长。
分别对甲醇-三乙胺-醋酸铵流动相、乙腈-四氢呋喃-醋酸铵的流动相进行考察,结果表明,以乙腈-四氢呋喃-醋酸铵为流动相时,所得到的色谱峰峰形好,分离效果好。故选择乙腈-四氢呋喃(V∶V=25 ∶15)作为流动相A,0.1 mol/L醋酸铵溶液(含冰醋酸0.05%)作为流动相B。
本研究建立了高效液相色谱法测定腰痛丸(Ⅰ)中新乌头碱、次乌头碱和乌头碱含量的方法,具有以下优势:(1)测定方法操作简便;(2)线性关系良好,测定方法精密度好;(3)结果准确可靠。
综上所述,本研究测定方法操作简便,分离效果佳,精密度、稳定性、重复性良好,为腰痛丸(Ⅰ)的质量控制提供了可靠的测定依据。
[1]国家药典委员会.中华人民共和国药典:一部[S].2015年版.北京:中国医药科技出版社,2015:39.
[2]任欣欣,李兴华,叶莎,等.HPLC测定复脉灵胶囊中乌头碱、新乌头碱、次乌头碱的含量[J].中国实验方剂学杂志,2013,19(18):131-134.
[3]李智勇,邓亚利,孙冬梅.HPLC法测定癌痛巴布剂中乌头碱、次乌头碱、新乌头碱[J].中成药,2011,33(6):988-991.
[4]罗霄,彭善贵,文永盛,等.HPLC测定制川乌中的乌头碱、次乌头碱和新乌头碱[J].华西药学杂志,2010,25(4):472-473.
[5]朱俊访,李卓亚,沈志滨,等.HPLC法测定不同地区制川乌中新乌头碱、次乌头碱及乌头碱的含量[J].食品与药品,2008,10(9):44-46.
[6]丘振文,罗丹冬,王沛坚.HPLC法测定舒痹宁颗粒中次乌头碱、新乌头碱的含量[J].中药新药与临床药理,2008,19(4):304-306.
[7]张宇洁,高苏亚,韩汾汾.反相高效液相色谱法同时测定乌头不同部位中新乌头碱和乌头碱含量[J].中南药学,2013,11(7):551-553.
[8]马芳,赵东,吴晓霞,等.LC-MS-MS法检测附子水提液中新乌头碱、次乌头碱的含量[J].中国实验方剂学杂志,2011,17(13):95-97.
[9]崔萍,杨莉,熊爱珍,等.UPLC-MS法测定消肿片中3个乌头碱类双酯型生物碱的含量[J].药物分析杂志,2012,32(5):868-872.
[10]林佶,杨艳国,张昆仑,等.HPLC快速测定草乌中毒患者尿液中的乌头碱、次乌头碱、新乌头碱[J].中国卫生检验杂志,2016,26(4):486-487,490.
[11]刘同梅,刘欣欣.HPLC法测定嘎日迪五味丸中的乌头碱、中乌头碱和次乌头碱的含量[J].现代中西医结合杂志,2012,21(19):2131-2132.
[12]Yang Y,Chen J,Shi YP.Determination of aconitine, hypaconitine and mesaconitine in urine using hollow fiber liquid-phase microext-raction combined with high-performance liquid chromatography[J].J Chromatogr B Analyt Technol Biomed Life Sci,2010, 878(28):2811-2816.
[13]Zhang CH,Wu HQ,Huang XL,et al.Simultaneous Determination of Toxic Alkaloids in Blood and Urine by HPLC-ESI-MS/MS[J].Chromatographia,2012,75:499-511.
[14]樊磊磊,杨艳玲,刘乃强,等.痹通药酒中新乌头碱、次乌头碱、乌头碱的含量[J].中国实验方剂学杂志,2012,18(22):98-100.
[15]刘敏,张海,蔡亚梅,等.附子总生物碱提取物中3个双酯型和3个单酯型乌头碱成分的含量测定[J].药学实践杂志,2013,31(3):181-184.
[16]卢永昌,薛二伟.HPLC测定黄毛翠雀花中的乌头碱、新乌头碱和次乌头碱[J].华西药学杂志,2014,29(2):206-207.
Content Determination of Mesaconitine, Hypaconitine and Aconitine in Yaotong Pills(I) by HPLC
ZHU Ganhong1,QIU Qiyuan2,CAO Guiping1,WU Shilong2
(1.Dept.of Parmacy, Yancheng Traditional Chinese Medicine Hospital, Jiangsu Yancheng 224001,China; 2.Dept.of Pharmaceutical Analysis,Yancheng Institute for Drug Control, Jiangsu Yancheng 224001,China)
OBJECTIVE:To establish the method for the content determination of mesaconitine, hypaconitine and aconitine in Yaotong pills(I), so as to further improve the quality control of Yaotong pills(I). METHODS: CAPCELL PAK C18(4.5 mm×250 mm,5 μm) column was adopted with the mobile phase A of acetonitrile- tetrahydrofuran (V∶V=25 ∶15) and the mobile phase B of 0.1 mol/L ammonium acetate solution by gradient elution at detection wavelength of 235 nm. The column temperature was 30 ℃ and the flow rate was 1.0 ml/min. RESULTS: The linearity ranges for mesaconitine, hypaconitine and aconitine were 0.2-1.6 μg/ml, the correlation coefficients werer2=0.999 1,r2=0.998 9,r2=0.999 5, respectively, and the average recoveries were 97.69% with relative standard deviation (RSD) 0.86%, 98.17% withRSD0.93%, 99.50% withRSD0.45%, respectively. CONCLUSIONS: The method is convenient, accurate with good repetition, which can be used for quality control of Yaotong pills (I).
HPLC; Yaotong pills(I); Mesaconitine; Hypaconitine; Aconitine
副主任药师。研究方向:药物分析、药物有效成分。E-mail:gita_80@126.com
R927.2
A
1672-2124(2016)09-1229-03
10.14009/j.issn.1672-2124.2016.09.030
2016-06-02)
*副主任药师。研究方向:临床药物分析、药事管理。E-mail:89695388@qq.com
