固相萃取-毛细管气相色谱法测定生活饮用水中16种硝基苯类化合物
2016-11-01邹海民周琛余辉菊张潜曾红燕李永新成都市疾病预防控制中心成都60047四川大学华西公共卫生学院成都6004
邹海民周 琛余辉菊张 潜曾红燕李永新(成都市疾病预防控制中心,成都 60047)(四川大学华西公共卫生学院,成都 6004)
固相萃取-毛细管气相色谱法测定生活饮用水中16种硝基苯类化合物
邹海民*1周 琛2余辉菊1张 潜1曾红燕2李永新21
(成都市疾病预防控制中心,成都 610047)2(四川大学华西公共卫生学院,成都 610041)
建立了生活饮用水中16种硝基苯类物质的固相萃取-毛细管气相色谱-电子捕获检测方法。实验优化了色谱柱、升温程序等色谱条件,并对影响固相萃取效果的主要因素(萃取小柱、洗脱剂和洗脱体积)进行了考察。水样中16种硝基苯类物质经Oasis HLB固相萃取柱吸附、正己烷-丙酮(3∶1,V/V)洗脱后,采用DB-1701毛细管气相色谱柱程序升温分离、ECD检测,在33 min内完成所有待测组分的测定。16种硝基苯类组分在各自的线性范围内相关系数r≥0.998,方法的检出限为0.01~0.77 μg/L(S/N=3),定量限为0.03~2.57 μg/L(S/N=10),日内精度和日间精度分别在1.0% ~3.8%和2.3% ~4.8%间,样品加标回收率为83.6%~111.8%,加标样品的RSD为1.2%~5.1%。应用本方法对50份水样进行了分析,结果表明,本方法准确、灵敏、快速,适用于水质的常规分析,可为水样中硝基苯类物质的污染评价提供技术支持。
气相色谱法;固相萃取;硝基苯类;饮用水
1 引言
硝基苯类化合物种类繁多,包括硝基苯、硝基氯苯、硝基甲苯等几大类,其结构稳定,难以降解。该类高毒物质可通过各种途径进入人体,造成消化、呼吸及神经系统等损害[1],并具有致突变性和致癌性[2,3]。美国EPA已将多种硝基苯列入“优先控制污染物”名单;我国《地表水环境质量标准》[4]中也对集中式生活饮用水地表水源中硝基苯类化合物的含量做出限定,其中硝基苯限值为0.017 mg/L;1-氯-2-硝基苯、1-氯-3-硝基苯和1-氯-4-硝基苯限值为0.05 mg/L;1,2-二硝基苯、1,3-二硝基苯、1,4-二硝基苯、1-氯-2,4-二硝基苯和2,4,6-三硝基甲苯限值均为0.5 mg/L。
目前,水质中硝基苯类化合物的检测多采用液相色谱法[5~7]、气相色谱法[8~10]和气相色谱-质谱联用法[11~13]等。液相色谱法分析时间过长;气相色谱-质谱联用法准确度好,但仪器设备较昂贵。毛细管气相色谱法在有机污染物分析方面具有快速、分离度好、准确度高,以及仪器条件要求相对较低等优点,已经被逐步应用于常规环境监测中。
《生活饮用水标准检验方法》[14]将硝基苯类物质分为硝基苯、三硝基甲苯、二硝基苯等几类进行测定,分别用不同溶剂进行液液萃取,且大部分采用不同填料的填充柱分离,采用电子捕获检测器(Electron capture detector,ECD)或火焰离子化检测器(Flame ionization detector,FID)进行检测。此法不仅前处理复杂、分离效能不高,且费时费力。本实验通过对固相萃取柱种类、萃取条件(洗脱液选择及配比)及色谱分离条件等进行优化,建立了固相萃取-毛细管气相色谱法,实现了对水中16种硝基苯类物质的同时检测。本方法灵敏度高、准确度好且简便快速,适合生活饮用水中硝基苯类化合物的同时检测。
2 实验部分
2.1 仪器与试剂
Varian cp-3800气相色谱仪,配有1171自动进样器、电子捕获检测器;固相萃取仪(美国Supelco公司);Oasis HLB固相萃取小柱(500 mg,6 mL,美国 Waters公司);Bond Elut Plexa固相萃取小柱(500 mg,6 mL,美国Agilent公司);纯水仪(PURELAB Pulse,德国ELGA公司);万分之一天平(德国Sartorius公司),HSC-24B氮吹仪(天津恒奥科技发展有限公司)。毛细管柱DB-1701(30 m×0.25 mm,0.25μm,Agilent J&W GC Columns)和DB-5MS(30 m×0.25 mm,0.25μm,Agilent J&W GC Columns)。
正己烷(n-Hexane)、丙酮(Acetone,Ac)、甲醇(色谱纯,美国Fisher Scientific公司)。
标准品:2-硝基甲苯(2-Nitrotoluene,2-NT)、3-硝基甲苯(3-Nitrotoluene,3-NT):浓度1.00 mg/L,环境保护部标准样品研究所;硝基苯(Nitrobenzene,NB):浓度1.00 mg/mL,中国计量科学研究所;4-硝基甲苯(4-Nitrotoluene,4-NT)、1-氯-4-硝基苯(1-Chloro-4-nitrobenzene,1-Ch-4-NB)、2,4-二硝基甲苯(2,4-Dinitrotoluene,2,4-DNT)、2,4,6-三硝基甲苯(2,4,6-Trinitrotoluene,TNT):浓度1.01 mg/L,中国计量科学研究所;1,2-二硝基苯(1,2-Dinitrobenzene,1,2-DNB):纯度99.9%,美国Sigma-Aldrich公司;1,3-二硝基苯(1,3-Dinitrobenzene,1,3-DNB)、1,4-二硝基苯(1,4-Dinitrobenzene,1,4-DNB)、2,5-二硝基甲苯(2,5-Dinitrotoluene,2,5-DNT):纯度99.9%,美国Supelco公司;3,4-二硝基甲苯(3,4-Dinitrotoluene,3,4-DNT)、1-氯-2-硝基苯(1-Chloro-2-nitrobenzene,1-Ch-2-NB)、1-氯-3-硝基苯(1-Chloro-3-nitrobenzene,1-Ch-3-NB)、1,3,5-三硝基苯(1,3,5-Trinitrobenzene,TNB):纯度99.9%,德国Dr.Ehrenstorfer公司;1-氯-2,4-二硝基苯(1-Chloro-2,4-dinitrobenzene,1-Ch-2,4-DNB):浓度100 mg/L,德国Dr.Ehrenstorfer公司。
2.2 试剂配制
2.2.1 单一标准储备液的配制准确称量各标准物质,分别用正己烷溶解并定容,配制成浓度为100.0 mg/L的单合标准储备液,4℃保存备用。
2.2.2 混合标准储备液的配制分别取适量标准储备液,用正己烷配制成混合标准储备液。其中NB,2-NT,3-NT,4-NT浓度为10.0 mg/L,1,3-DNB,TNB浓度为2.0 mg/L,其余(2,4-DNT,TNT,2,5-DNT,3,4-DNT,1,2-DNB,1,4-DNB,1-Ch-2-NB,1-Ch-3-NB,1-Ch-4-NB,1-Ch-2,4-DNB)浓度为0.50 mg/L,4℃保存备用。
2.3 实验方法
2.3.1 GC-ECD色谱条件进样口温度250℃;载气为高纯氮气,流速为2 mL/min;柱温箱升温程序:初始温度60℃保持1 min,以4℃/min升温至140℃后,再以15℃/min升温至240℃,并保持5 min;检测器温度300℃;尾吹气(N2)流速60 mL/min;进样体积:1 μL,不分流。
2.3.2 样品前处理量取500 mL水样于烧杯中,加入2.5 mL甲醇,混匀,以5 mL/min流速经HLB固相萃取柱(预先依次用5 mL正己烷、5 mL甲醇、5 mL纯水活化)。然后用10 mL纯水淋洗,弃去淋洗液,再用10 mL正己烷-丙酮(3∶1,V/V)以2 mL/min流速进行洗脱,洗脱液经干燥柱除水后,收集于接收管中,并定容至10 mL,待测。取500 mL实验用超纯水代替水样,同样进行上述操作,作为样品空白。
3 结果与讨论
3.1 色谱条件优化
3.1.1 色谱柱的选择本研究比较了HP-5MS和DB-1701两种色谱柱对16种硝基苯类物质的分离效果。如图1所示,DB-1701对难分离组分1-Ch-3-NB,1-Ch-4-NB和1-Ch-2-NB分离效果更好,且峰型比HP-5MS条件下更好,但1-Ch-3-NB和4-NT的分离度比HP-5MS条件下小,因此实验选择DB-1701毛细管柱作为分离柱,并进一步进行分离条件优化。
3.1.2 柱温的选择由于16种待测硝基苯类化合物的沸点相差较大,沸程长,采用恒温模式无法达到基线分离,故本研究选用程序升温模式。
本研究首先考察了初始温度分别为40℃,60℃,80℃和100℃时16种待测物质的分离情况(以4℃/min升温至140℃,再以15℃/min升温至240℃,并保持5 min),结果(图2)表明,随着初始温度升高,组分的保留时间缩短,分析周期变短,但是分离度变差,特别是4-NT和1-Ch-3-NB分离度明显下降。当初始温度为40℃时,4-NT与1-Ch-3-NB能达到基线分离,但整个分析过程所需时间过长(42 min);当初始温度为60℃时,4-NT与1-Ch-3-NB也能达到基线分离,同时硝基苯及硝基甲苯等前7种物质峰型较好,且分析时间缩短至33 min;初始温度升高至80℃时,4-NT与1-Ch-3-NB已经无法实现基线分离,且随着初始温度的升高,硝基苯、硝基甲苯、硝基氯苯等前7种物质出现不同程度的峰型展宽。因此实验选择分析时间较短且能实现各物质基线分离的60℃作为最佳初始温度。

图1 不同毛细管柱上16种硝基苯色谱图Fig.1 Chromatograms of 16 nitrobenzene compounds separated by two columns

图2 初始柱温的影响Fig.2 Effect of different initial column temperatures
固定初始温度为60℃,考察了第一阶段升温速率分别为4℃/min,6℃/min及8℃/min时(升至140℃,15℃/min,升温至240℃,并保持5 min),待测物质的分离情况。结果显示:随着升温速率的增大,分析时间逐渐缩短,但分离度也明显降低。当升温速率超过4℃/min时,4-NT与1-Ch-3-NB未达到基线分离,故选择阶段升温速率为4℃/min(见图3)。

图3 程序升温速率的影响Fig.3 Effect of different temperature programs
3.2 样品前处理条件优化
3.2.1 固相萃取柱的选择根据待测物的性质,本实验初步选择C18固相萃取小柱和HLB固相萃取小柱进行水样前处理。取500 mL超纯水6份,分别加入相同浓度混合标准溶液,匀,其中3份采用C18萃取小柱萃取,另3份采用HLB固相萃取小柱进行萃取,通过考察平均回收率来评价这两种小柱对待测物的萃取效率。结果(见图4)显示,HLB固相萃取小柱除了对NB和3-NT的萃取效率略低于C18柱外,对其它14种待测物的萃取效率均明显高于C18柱。综合考虑,最终选用HLB固相萃取小柱。
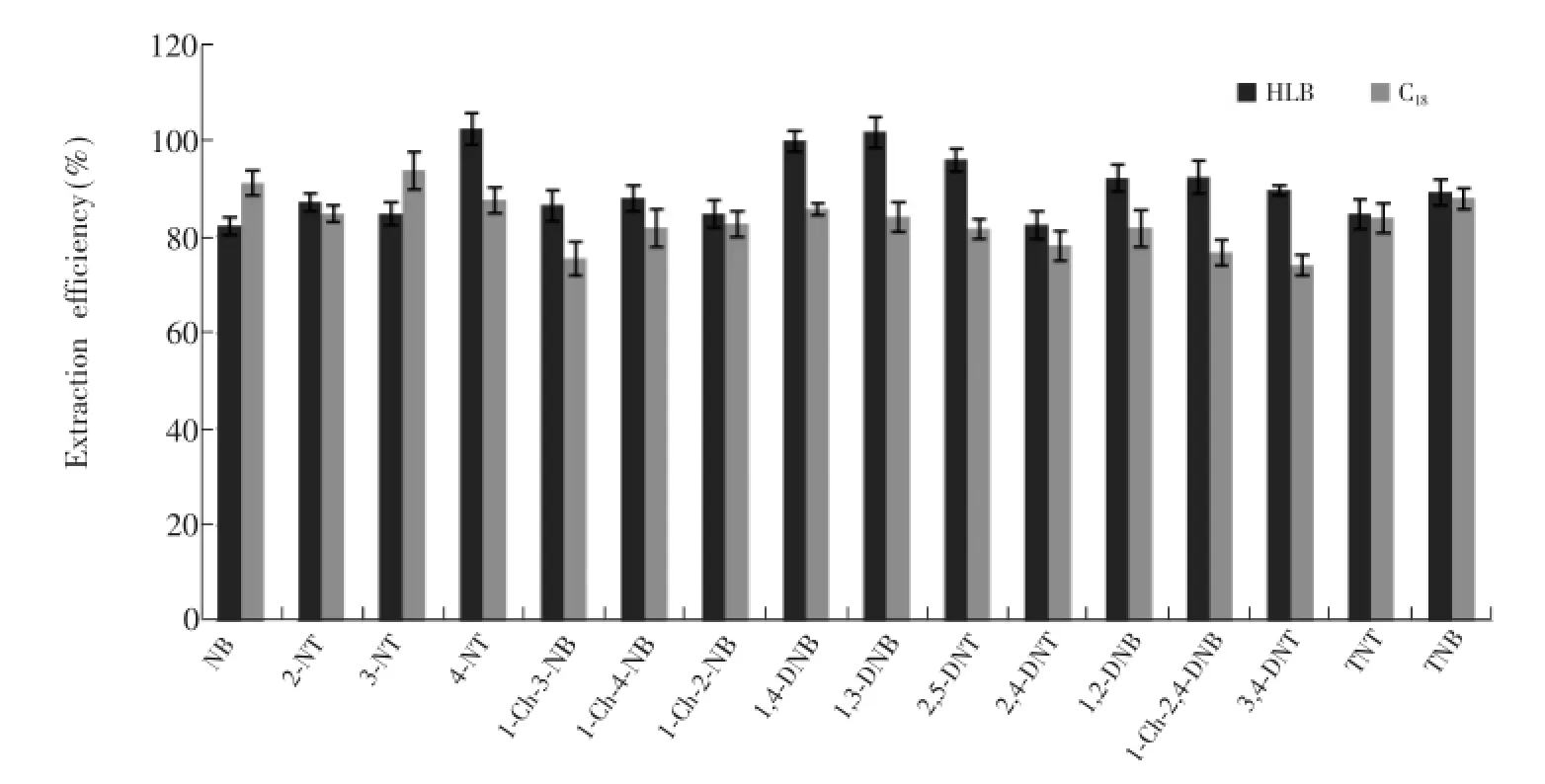
图4 不同萃取柱的萃取效率Fig.4 Extraction efficiency of two extraction columns
3.2.2 洗脱剂的选择常用的洗脱剂有乙腈[12]、二氯甲烷[16]、正己烷[17]、丙酮[18]等,其中正己烷和丙酮最为常用。文献[19]报道,以一定比例混合的正己烷和丙酮作为洗脱剂对硝基苯类物质的洗脱效率更高,本实验考察了洗脱剂分别为正己烷、丙酮及两者混合比例(正己烷-丙酮体积比依次为4∶1,3∶1,2∶1,1∶1)时的洗脱结果(见图5)。结果表明,单独使用正己烷和丙酮时,洗脱效果明显不如混合洗脱剂,当正己烷-丙酮的体积比为3∶1时,洗脱效果最好,故选择正己烷-丙酮(3∶1,V/V)作为最佳洗脱试剂。
3.2.3 洗脱剂体积的选择保持其它实验条件不变,改变洗脱试剂体积(5,7.5,10,12.5和15 mL),比较其加标回收率。结果表明,随着洗脱剂体积的增大,组分的洗脱效果变好,当洗脱剂体积为10 mL,回收率为93.5%,继续增加洗脱剂体积,洗脱效果不变。故选择洗脱剂最佳洗脱体积为10 mL。
图5 不同洗脱剂的洗脱效果Fig.5 Elution efficiency of different eluents
3.3 方法学评价
3.3.1 回归方程、线性范围和检出限分别吸取1 μL标准系列溶液,按2.3.1节的色谱条件分别进样分析,每个浓度重复测定3次,以待测物浓度(x,μg/L)为横坐标,峰面积平均值(y,mV·min)为纵坐标,绘制标准曲线。以3倍和10倍基线噪声所对应的各组分含量计算方法检出限(Limit of detection,LOD)和定量限(Limit of quantification,LOQ)。结果(表1)表明,在各自的线性范围内,16种硝基苯类组分相关系数r≥0.998,方法的检出限为0.01~0.77 μg/L(S/N=3),定量限为0.03~2.57 μg/L (S/N=10)。而《生活饮用水标准检验方法》(GB/T 5750-2006)中最低检测质量浓度(取500 mL水样)分别为:1-Ch-3-NB,1-Ch-4-NB,1-Ch-2-NB为20 μg/L;1,4-DNB为40 μg/L;1,2-DNB,1-Ch-2,4-DNB为100 μg/L,1,3-DNB为200 μg/L。因此,本实验所建立的方法能够满足实际检测对检出限的要求。

表1 线性范围、回归方程、相关系数、检出限及定量限Table 1 Linear range,regression equation,correlation coefficient,LOD and LOQ
3.3.2 精密度取同一水源水,加入16种组分的混合标准溶液作为模拟样品进行精密度实验,按2.3.1节的条件进行测定,在同一天内连续进样6次,以峰面积和保留时间的相对标准偏差(Relative standard derivation,RSD)考察日内精密度;连续6天进样测定,考察日间精密度。结果(见表2)显示,日内精度和日间精密度分别在1.0%~3.8%和2.3%~4.8%之间,满足定量分析的基本要求。
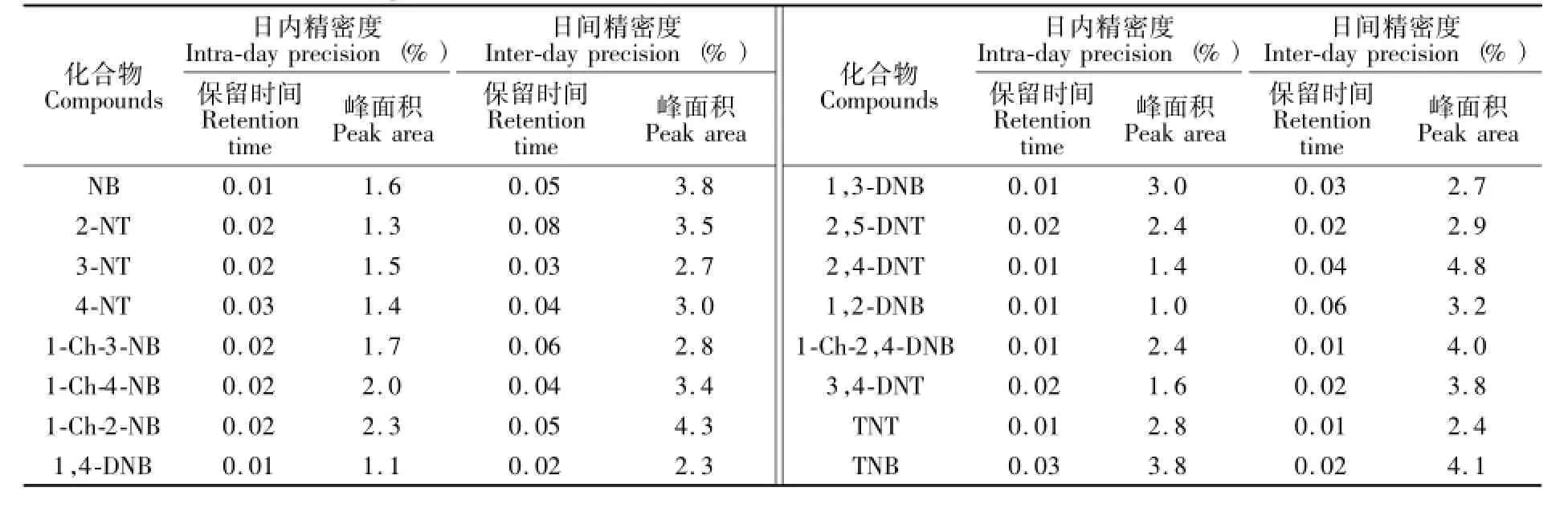
表2 日内精密度和日间精密度(n=6)Table 2 Intra-and inter-day precision(n=6)
3.4 加标回收实验
选取同一水源水分别加入3个浓度水平的标准溶液,结果见表3,平均回收率在83.6%~111.8%(n=6)之间,加标样品的RSD为1.2%~5.1%,表明本方法可用于检测生活饮用水中硝基苯类物质。
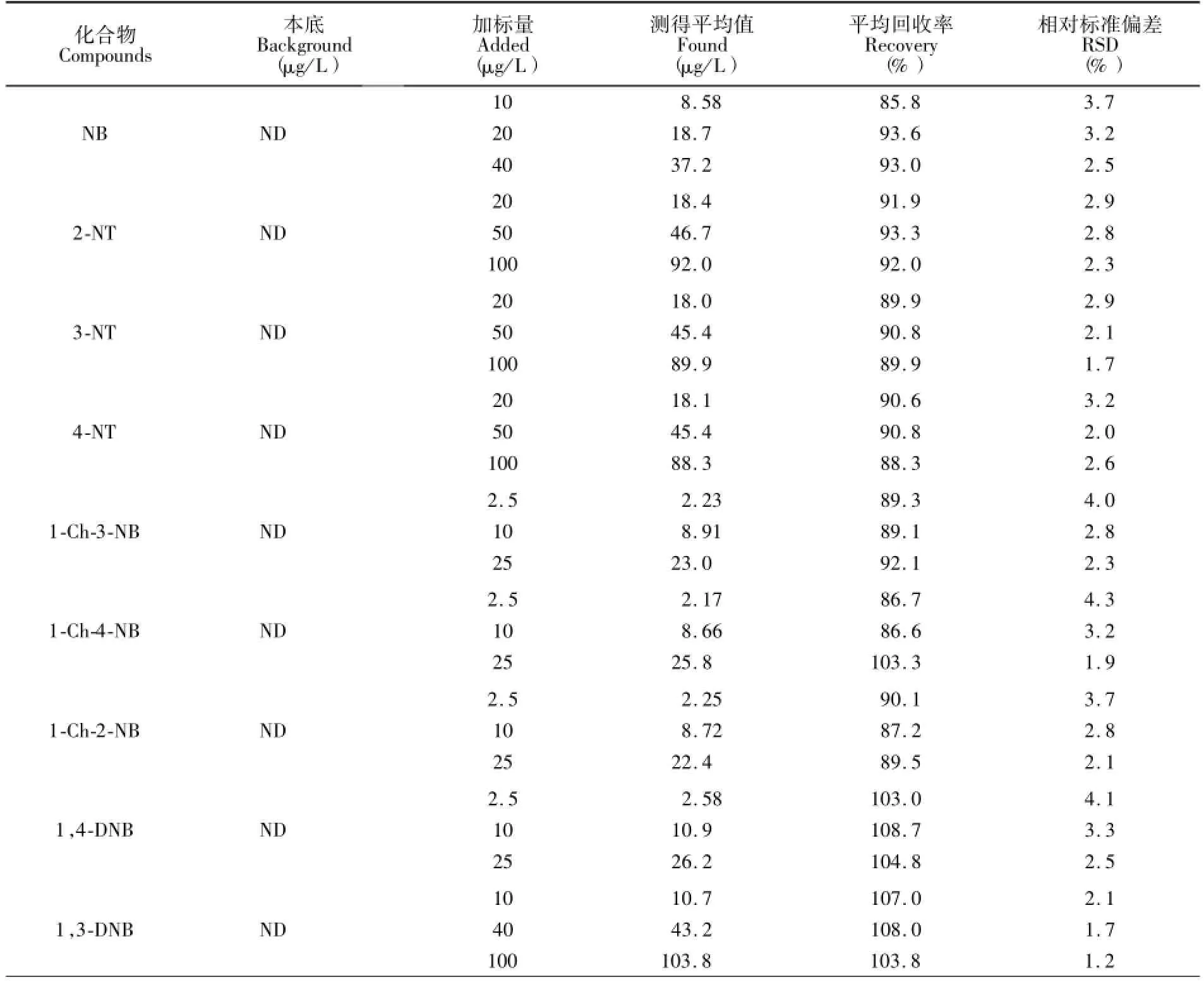
表3 加标回收率(n=6)Table 3 Results of the spiked recovery tests(n=6)

续表3(Continued to Table 3)
3.5 样品分析
应用本方法对成都市生活饮用水日常监测水样进行检测,其中水源水10份(经0.45μm水系滤膜过滤)、出厂水20份、末梢水20份,每份样品均做3个平行样,结果表明,50份样品均未检出16种待测物质。于是考虑对样品进行加标做模拟样,取水源水、出厂水和末梢水各10份,按2.3.3节中表3进行3个浓度加标实验。结果表明,在该水样基质条件下各物质的保留时间变化不大,且16种待测物质的回收率均在78.6%~115.3%之间。与GB/T 5750.8-2006《生活饮用水标准检验方法有机物指标》采用传统的液液萃取,GC-FID、GC-ECD分组测定方法相比,本方法能实现一次进样在33 min内实现16种硝基苯的同时检测,方法简便、快速、准确、灵敏度高,适于生活饮用水中硝基苯类化合物的检测。
1 Kovacic P,Somanathan R.J.Appl.Toxicol.,2014,34(8):810-824
2 Imamura T,Koeda A,Morimoto K,Hatakeyama H,Suzuki H,Wako Y,Kawasako K,Otabe K,Sato S.Mutat.Res-gen. Tox.En.,2015,780-781:46-50
3 Oh J H,Heo S H,Park H J,Choi M S,Lee E H,Park S M,Nam Y S,Yoon S.Reprod.Toxicol.,2014,43:45-55
4 GB 3838-2002 Environmental quality standards for surface water.National Standards of the People′s Republic of China
地表水环境质量标准.中华人民共和国国家标准.GB 3838-2002
5 LIU Peng,ZHANG Lan-Ying,JIAO Yan-LIN,LIU Na,LIU Ying-Ying,GAO Song.Chinese J.Anal Chem.,2009,37(5):741-744
刘鹏,张兰英,焦雁林,刘娜,刘莹莹,高松.分析化学,2009,37(5):741-744
6 HUANG Yi,RAO Zhu,LIU Yan,LIU Chen,GOU Xiao-Chen.Rock and Mineral Analysis,2012,31(04):666-671
黄毅,饶竹,刘艳,刘晨,郭晓辰.岩矿测试,2012,31(4):666-671
7 Wang S P,Chen H J.J.Chromatogr.A,2002,979(1):439-446
8 Li X,Chen J M,Du L.J.Chromatogr.A,2007,1140(1):21-28
9 Ebrahimzadeh H,Yamini Y,Kamarei F,Zanjani M K.Talanta,2007,72(1):193-198
10 Guan W,Xu F,Liu W M,Zhao J H,Guan Y F.J.Chromatogr.A,2007,1147(1):59-65
11 Zhang G J,Zhou X,Zang X H,Li Z,Wang C,Wang Z.Chinese Chem.Lett.,2014,25(11):1449-1454
12 Jönsson S,Gustavsson L,Bavel BV.J.Chromatogr.A,2007,1164(1):65-73
13 Wang L,Wang L L,Chen J,Du W J,Fan G L,Lu X H.J.Chromatogr.A,2012,1256:9-14
14 GB/T 5750-2006 Standard Examination Methods for Drinking Water.National Standards of the People′s Republic of China
生活饮用水标准检验方法.中华人民共和国国家标准.GB/T 5750-2006
15 Cao X J,Shen L X,Ye X M,Zhang F F,Chen J Y,Mo W M.Analyst,2014,139(8):1938-1944
16 HJ 648-2013 Water Quality-Determination of Nitroaromatics by Gas Chromatography.National Environmental Protection Standards of the People′s Republic of China.
水质硝基苯类化合物的测定液液萃取/固相萃取-气相色谱法.中华人民共和国国家环境保护标准.HJ 648-2013
17 Parham H,Saeed S.J.Ind.Eng.Chem.,2014,20(3):1003-1009
18 LI Li-Rong,WEI En-Qi,SHI Ting-Rui,WANG Yan-Li,YANG Hua.Physical Testing and Chemical Analysis(Part B),2011,(12):1436-1439
李利荣,魏恩棋,时庭锐,王艳丽,杨华.理化检验-化学分册,2011,(12):1436-1439
19 Sautilio A,Ziemacki G,Stefanelli P,Dommarco R.J.Environ.Sci.Health A Tox.Hazard.Subst.Environ.Eng.,2008,43:437-442
This work was supported by the Science&Technology Supporting Program of Sichuan Province,China(No.2012SZ0196)
Simultaneous Determination of 16 Nitrobenzene Compounds in Drinking Water by Solid Phase Extraction-Gas Chromatography coupled with Electron Capture Detection
ZOU Hai-Min*1,ZHOU Chen2,YU Hui-Ju1,ZHANG Qian1,ZENG Hong-Yan2,LI Yong-Xin21(Chengdu Center for Disease Control&Prevention,Chengdu 610047,China)
2(West China School of Public Health,Sichuan University,Chengdu 610041,China)
A solid phase extraction(SPE)-gas chromatography coupled with electron capture detection(GCECD)method was developed for the simultaneous determination of 16 nitrobenzene compounds in drinking water.The chromatographic conditions,i.e.the chromatographic column and the programmed temperature,were optimized,and the extraction conditions,i.e.the type of extraction column,elution solvent and volume of elution solvent,were also optimized.Water samples were extracted with Oasis HLB columns and eluted by the mixture of n-hexane and acetone(3∶1,V/V).All the target compounds were chromatographically separated on a DB-1701 capillary column with programed temperature,and were detected by electron capture detector.The favorable resolutions of all target compounds were achieved within 33 min.Under the optimal analytical conditions,the peak area of each analyte and its concentration had a good correlation within the linear range(r≥0.998).The limit of detection(LOD)and limit of quantification(LOQ)of the method were 0.01-0.77 μg/L(S/N=3)and 0.03-2.57 μg/L(S/N=10),respectively.The intra-and inter-day relative standard deviations(RSDs)of the sample were 1.0%-3.8%and 2.3%-4.8%,respectively. Spiked recoveries of the analytes were 83.6%-111.8%and the RSDs of the spiked samples were 1.2%-5.1%.This proposed method has been applied in the detection of 50 water samples.The results indicated that the new solid phase extraction-gas chromatography coupled with electron capture detection method was specific,sensitive and reproducible,and could be used for the determination of 16 nitrobenzene compounds in drinking water.
Gas chromatography;Solid phase extraction;Nitrobenzene;Drinking water
15 August 2015;accepted 22 October 2015)
10.11895/j.issn.0253-3820.150648
2015-08-15收稿;2015-10-22接受
本文系四川省科技支撑计划基金(No.2012SZ0196)资助项目
*E-mail:742779591@qq.com
