R基团搜索技术用于HIV-1逆转录酶抑制剂的分子设计
2016-10-16仝建波
仝建波, 白 敏, 赵 翔, 常 佳
(陕西科技大学化学与化工学院,陕西科技大学教育部轻化工助剂化学与技术重点实验室,陕西西安 710021)
艾滋病(Acquired Immunodeficiency Syndrome,AIDS)是由人类免疫缺陷病毒(Human Immunodeficiency Virus,HIV)感染引起的全身性传染病[1]。HIV可分为Ⅰ型(HIV-1)和Ⅱ型(HIV-2)[2,3],二者之间存在一定的免疫交叉反应。艾滋病绝大多数由HIV-1引起。逆转录酶(Reverse Transcriptase,RT)在HIV-1的复制循环中起着关键性的作用,因此,HIV-1逆转录酶是抗HIV/AIDS药物发展中的一个重要生物靶点[4]。针对HIV-1逆转录酶的药物分为核苷类逆转录酶抑制剂(Nucleoside Reverse Transcriptase Inhibitors,NRTIs)和非核苷类逆转录酶抑制剂(Non-nucleoside Reverse Transcriptase Inhibitors,NNRTIs)[5]。NNRTIs类药物具有活性高、毒性低、结构多样化、酶的结构清楚、药物作用靶点明确等优点[6 - 8],因此一直是寻找新的抗艾滋病药物的重要方向之一。
定量构效关系(QSAR)作为一种重要的药物设计方法,通常来讲有两种QSAR模型:2D-QSAR和3D-QSAR。CoMFA[9]是3D-QSAR中应用比较广泛的一种方法。本文采用第二代CoMFA方法,即Topomer CoMFA[10,11]方法对41个HIV-1逆转录酶抑制剂进行3D-QSAR分析。所建立的Topomer CoMFA模型结合Topomer Search[12]技术可以进行基于配体的虚拟筛选,为新药的设计奠定基础。不同于CoMFA的是:Topomer CoMFA是通过一系列完全客观一致的叠合规则完成3D-QSAR分析的准备工作,并且具有重复性高的优势。利用Topomer CoMFA方法可以快速建立预测模型并进行分析与评价,为同类小分子抑制剂的结构优化提供理论依据[13]。
1 方法与步骤
1.1 数据集选择与划分
41个HIV-1逆转录酶抑制剂分子结构与活性数据见表1[14]。活性标度为pIc50(-logIc50),其中Ic50值为HIV-1病毒感染细胞半数抑制浓度。按照随机化原则将数据集划分为36个训练集分子和5个测试集分子,训练集用来建立模型,测试集用以验证模型的外部预测能力。此外,对13号和34号分子的不同构象进行了验证,结果见表1。
表141个HIV-1逆转录酶抑制剂的结构及其pIc50
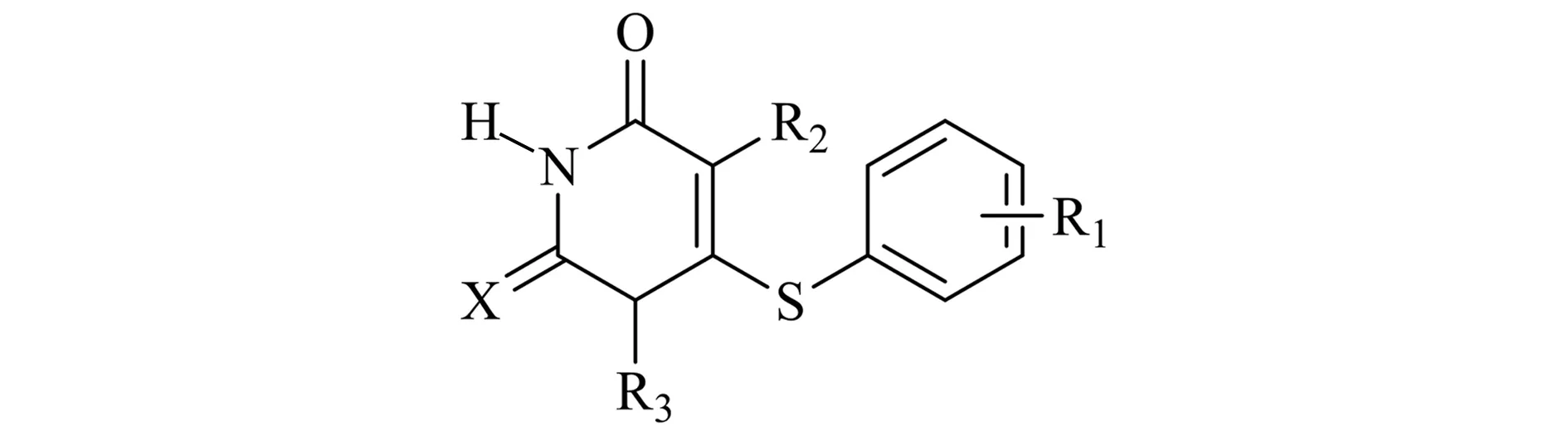

No.R1R2R3XExperimental pIc50Predicted pIc5013-CF3MeCH2OCH2CH2OHO4.354.3123-FMeCH2OCH2CH2OHO5.485.4033-ClMeCH2OCH2CH2OHO4.895.0143-BrMeCH2OCH2CH2OHO5.245.1253-IMeCH2OCH2CH2OHO5.005.0563-NO2MeCH2OCH2CH2OHO4.474.5273-OHMeCH2OCH2CH2OHO4.094.1383-OMeMeCH2OCH2CH2OHO4.664.6593,5-Me2MeCH2OCH2CH2OHO6.596.71103,5-Cl2MeCH2OCH2CH2OHO5.896.0711Hi-PrCH2OCH2CH2OHO7.207.42123,5-Me2EtCH2OCH2CH2OHO7.897.52133,5-Me2i-PrCH2OCH2CH2OHO8.578.50,8.3914HMeCH2OCH2PhO7.067.0115HEtCH2OCH2MeO7.727.5816HEtCH2OCH2MeS7.587.4517a3,5-Me2EtCH2OCH2MeO8.248.67183,5-Me2EtCH2OCH2MeS8.308.5419HEtCH2O-c-HexS5.795.6920HEtCH2OCH2-c-HexS6.456.4321HEtCH2OCH2C6H4(4-Me)S7.117.1822HEtCH2OCH2C6H4(4-Cl)S7.927.9023HEtCH2OCH2CH2PhS7.047.09243,5-Cl2EtCH2OCH2MeO8.138.0325HEtCH2O-i-PrO6.476.5626aHEtCH2O-c-HexO5.405.7527HEtCH2OCH2-c-HexO6.356.4728aHEtCH2OCH2CH2PhO7.047.11294-NO2MeCH2OCH2CH2OHO3.723.6830a4-CNMeCH2OCH2CH2OHO3.603.34314-OHMeCH2OCH2CH2OHO3.563.44324-OMeMeCH2OCH2CH2OHO3.603.64334-COMeMeCH2OCH2CH2OHO3.963.96344-COOHMeCH2OCH2CH2OHO3.453.48,3.38353-CONH2MeCH2OCH2CH2OHO3.513.5336HCOOMeCH2OCH2CH2OHO5.185.1637aHCONHPhCH2OCH2CH2OHO4.744.1338HSPhCH2OCH2CH2OHO4.684.6739HCCHCH2OCH2CH2OHO4.744.6740HCCPhCH2OCH2CH2OHO5.475.52413-NH2MeCH2OCH2CH2OHO3.603.61
aChosen as the test set.
1.2 Topomer CoMFA模型建立

图1 13号分子的切割方式Fig.1 Cutting style of molecule 13
采用Sybyl 2.0-X软件包中的sketch molecule模块绘制出化合物的结构,对所有的化合物分子进行优化,优化过程采用Powell能量梯度法、Tripos力场,能量收敛设为0.05 kcal/mol,优化次数设定为1 000次,分子荷载电荷为Gasteiger-Huckel电荷,其余参数以Sybyl 2.0-X默认。
以活性最高的13号分子为模板对36个训练集化合物的结构进行切割,切割方式如图1。选择以S为中心进行切割生成Ra和Rb基团,接着软件会对其余的分子结构自动识别并进行切割,最后对Ra和Rb基团周围的立体场和静电场进行计算,并采用偏最小二乘回归分析法[15]建模生成3D-QSAR模型。采用留一法交互验证评价模型的内部预测能力。利用建立的Topomer CoMFA模型对5个测试集化合物的活性进行预测,以此评价模型的外部预测能力。
1.3 Topomer Search
采用Topomer Search技术可以从大量的化合物数据库中筛选出R基团(R-groups)。本文利用建立的Topomer CoMFA模型对ZINC(2012)数据库中的Drug-like类(包含130 000个分子)进行筛选,以得到具有高活性贡献的Ra和Rb基团,其中Topomer距离值设置为185,其他参数以Sybyl 2.0-X默认。
2 结果与讨论
2.1 Topomer CoMFA建模结果与评价

图2 41个抑制剂生物活性的实验值与预测值的线性回归图Fig.2 Linear regression between experimental and predicted pIC50of 41 inhibitors

2.2 Topomer CoMFA 等势图分析

图3 Topomer CoMFA模型三维等势图Fig.3 3D contour of Topomer CoMFA model(a)steric field map of Ra;(b)electrostatic field map of Ra;(c)steric field map of Rb;(d)electrostatic field map of Rb.
以13号分子为模板,Topomer CoMFA三维等势图见图3。图3(a)和3(c)分别表示Ra和Rb基团的立体场等势图,绿色区域表示引入体积较大的取代基有利于活性的提高,黄色区域表示不宜引入体积较大的取代基。图3(b)与3(d)分别表示Ra和Rb基团的静电场等势图,红色区域表示引入负电性取代基有利于活性的提高,蓝色区域表示引入正电性取代基有利于活性的提高。图3(a)中,R3取代基链端有大片黄色区域,R2取代基链端有大片绿色区域。图3(b)中R3取代基近端有大块蓝色和红色区域,R2取代基周围有大块蓝色区域。图3(c)中苯环的3位点和4位点处有大片黄色区域,5位点处有大块绿色区域。图3(d)中苯环的3位点、4位点和5位点处有大块红色区域。
综上所述,Ra基团的R3取代基处不适宜引入体积大的基团,例如以-CH2OCH2CH2OH取代17号分子的-CH2OCH2Me得到12号分子后活性降低。R2取代基处适宜引入体积较大的基团,例如以体积较大的异丙基取代12号分子的乙基得到13号分子后活性明显增大。Rb基团中苯环的3位和4位点处适宜引入体积较小并带负电的基团,5位点处适宜引人体积较大并带负电的基团,例如2号分子在此处引入体积小并带负电的氟基,而7号分子引入体积比氟基大并带正电的羟基,因此2号分子的活性大于7号分子。31、32号分子的活性较低是因为在苯环的4位点处引入了体积大的羟基和甲氧基。
2.3 Topomer Search
Topomer Search的结果中主要包含两项:Topomer距离和R基团的活性贡献值。Topomer距离通过计算值评价化合物与提问结构基团的结构相似性程度;而R基团的活性贡献则是基于Topomer CoMFA模型对R基团活性值的预测打分。基于Topomer CoMFA模型,采用Topomer Search技术对ZINC(2012)数据库进行筛选,得到5个Ra基团和5 000个Rb基团,以活性最高的13号分子为模板进行过滤,挑选出明显高于13号分子的Ra和Rb贡献值的R基团。最终得到1个Ra基团和20个Rb基团。
2.4 分子设计
用最终筛选得到的Ra和Rb基团分别替换13号分子的Ra和Rb基团,得到20个新化合物分子的结构,对新分子进行优化,优化方法同41个样本分子,利用建立的Topomer CoMFA模型对新化合物进行活性预测,结果有19个新设计的化合物预测活性值高于13号分子。新设计的分子Ra附近取代基团体积增大,因此活性有所增大。另外,化合物的活性高低还会受Rb基团影响。
3 结论
本文采用Topomer CoMFA方法对36个训练集HIV-1逆转录酶抑制剂进行了三维定量构效关系分析,并利用所建立的模型对5个测试集抑制剂进行了活性预测。采用Topomer Search技术对ZINC数据库进行R基团的虚拟筛选,并设计出20个具有更高活性的抑制剂分子。结果表明利用Topomer CoMFA方法能够建立有效的3D-QSAR模型,可以对未知化合物的生物活性进行预测,利用Topomer Search可以有效的从数据库中筛选出有高贡献的R基团,用以设计出新的药物分子,为抗艾滋病新药设计提供了新的候选物。
