梯恩梯/萘共晶初始热分解的反应分子动力学模拟
2016-10-14刘海何远航
刘海,何远航
(北京理工大学爆炸科学与技术国家重点实验室,北京100081)
梯恩梯/萘共晶初始热分解的反应分子动力学模拟
刘海,何远航
(北京理工大学爆炸科学与技术国家重点实验室,北京100081)
ReaxFF反应力场在冲击起爆、爆轰等问题中的应用多围绕常规含能材料。利用增加了长程修正项Elg的ReaxFF/lg反应力场对梯恩梯/萘(TNT/C10H8)共晶初始高温热分解进行模拟,并通过TNT单晶比较了C10H8对共晶整体反应特征的影响,利用指数函数及反应速率方程拟合得到共晶初级吸热反应和次级放热反应的活化能分别为35.7 kcal/mol和56.1 kcal/mol.初级吸热反应的活化能与TNT单晶基本相同,而次级放热反应的活化能则远远高于TNT单晶,并且同温度条件下共晶次级反应放热量小于TNT单晶。拟合得到的反应物的衰减速率表明,C10H8的加入将抑制共晶内TNT的分解。产物识别分析显示初始产物为NO2、NO和HONO,并且通过NO2和TNT—NO2,NO和TNT—NO,HONO和TNT—HONO分布数量的比较论证了固相共晶和TNT单晶的初始反应路径为双分子反应机制。共晶热分解最终主要产物为N2,H2O,CO2和CO.并且由于共晶内C10H8分子C—C键断裂所需的能量高于C—H键断裂所需的能量,C10H8分子的初始热分解路径为C—H键断裂,并且形成的H原子将促进共晶内H2O的产率高于TNT单晶内H2O的产率。
兵器科学与技术;共晶;高温热分解;反应动力学;ReaxFF反应力场
0 引言
极端条件下固相含能材料的物理化学响应问题是爆轰学的核心内容之一,并且,近几十年的实验测试和理论分析多围绕常规含能材料冲击起爆和爆轰波的宏观问题,如爆压、爆速、爆轰波结构以及稳定性等[1]。极端温度/压力条件下固相含能材料的初始反应路径是研究冲击起爆问题的基础,同样也与含能材料爆轰波能量输出结构直接关联。由于固相含能材料的冲击起爆及爆轰问题涉及极短的时间和极小的空间尺度,这给现有的实验技术带来了很大的挑战。近期,各国学者利用美国加州理工学院Goddard小组提出的ReaxFF反应力场分子动力学方法[2]已经就常规含能材料高温热分解的初始反应路径[3-5]以及冲击起爆机制[6-16]进行了广泛而深入地研究。
包含氧化剂和燃料组分的含能材料主要通过激发后释放的化学能产生高温/高压燃烧产物以达到毁伤、推进等效果。但限于冲击感度、密度、熔点、稳定性等因素,当前仅部分含能材料可用于战斗部和推进剂。共晶,作为一种改性技术,主要通过分子间非共价键将不同种类的含能材料分子结合在同一晶格中以实现含能材料的高能低感特性[17]。国内外在共晶含能材料领域的研究尚处于初步的实验室合成阶段,但已经取得了一定的成果(六硝基六氮杂异伍兹烷/梯恩梯(CL20/ TNT)[18-19]、六硝基六氮杂异伍兹烷/奥克托今(CL20/HMX)[20]、六硝基六氮杂异伍兹烷/三氟甲基苯(CL20/BTF)[21]、奥克托今/三氨基三硝基苯(HMX/TATB)[22]、梯恩梯/萘(TNT/C 10H8)[23]),实验结果业已表明上述共晶含能材料在爆速及安定性方面具有优异表现。当前,分子动力学技术在热分解以及冲击起爆等问题中的应用多面向常规含能材料,针对共晶含能材料的相关研究却鲜有发表[24-26]。本文利用ReaxFF反应力场分子动力学方法对TNT/C10H8共晶的初始热分解问题进行了研究,此结果可为优化共晶含能材料内分子配比、合成高能低感新型共晶含能材料等问题提供参考。
1 模型及计算细节
TNT/C10H8共晶初始单胞结构来自X射线衍射测试结果[23]。初始单胞内含有2个 TNT和2个C10H8分子。随后构建4×2×2共晶超胞(见图1),晶胞内TNT和C10H8的分子数量均为32个,共计1 248个原子。共晶初始密度为1.495 g/cm3,其中C10H8的密度为0.539 g/cm3.首先利用共轭梯度法通过系统势能收敛对超胞进行能量最小化。随后利用Berendsen热浴方法对超胞进行温度和压力弛豫,以获得常温(300 K)下的晶体结构,此过程持续5 ps.为了比较共晶内C10H8对TNT热分解的影响,相应地构建2×2×2正交TNT超胞结构(见图1),TNT初始单胞结构来自剑桥晶体结构数据库(CCDC)[27],初始密度为1.704 g/cm3,初始单胞内含有8个TNT分子。为了消除密度差异对热分解结果造成的影响,在构建TNT超胞前,对TNT单胞进行体积放大以获得近似于TNT/C10H8共晶的密度(1.510 g/cm3),超胞内共计64个TNT,1 344个原子。能量最小化以及温度和压力弛豫过程同上。最后对两含能材料晶体施加2 000 K~4 500 K高温以模拟含能材料爆轰Chapmam-Jouguet温度,此过程持续25 ps,整个模拟过程的时间步长为0.1 fs,高温热分解过程采用集成于lammps程序[28-29]内的ReaxFF/lg[30]分子动力学进行模拟。
ReaxFF/lg是在ReaxFF势函数的基础上增加了长程修正项Elg.ReaxFF/lg势函数在含能材料晶体结构以及状态方程的预测相比于ReaxFF势函数更加接近实验值[30]。当前,ReaxFF/lg势函数已经在含能材料感度分析[31]、热分解[24]、热冲击[32]、冲击压缩[33]、冲击起爆[6-7]等问题得到应用。
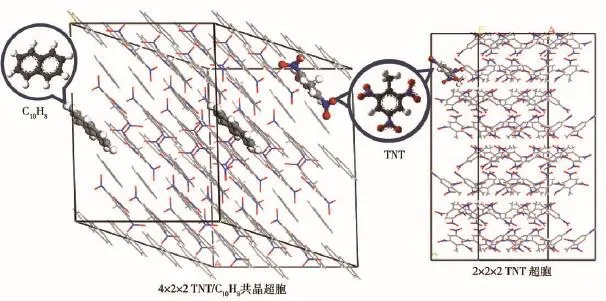
图1 TNT/C10H8共晶和TNT超胞模型Fig.1 Models of TNT/C10H8cocrystal and TNT supercell
2 结果及分析
2.1势能演化及曲线拟合
含能材料高温热分解过程中,系统将首先吸收热量使得系统势能(PE)升高,至峰值后,系统势能将随着放热化学反应的进行而开始衰减[3-5]。图2 为TNT/C10H8共晶和TNT高温热分解过程中系统势能随时间的演化曲线。当T=2 000 K时,TNT/ C10H8共晶和TNT分子吸收热量后,在有限的时间内并没有发生放热化学反应。当外界温度升至3 000 K以上,两种含能材料在经过短暂的“平衡-诱导期”后,系统势能开始衰减,且随着温度的升高,系统势能开始衰减的时刻逐渐提前,并且衰减速率逐渐增加,这表明温度的增加将促进放热化学反应的进行。比较同温度条件下两种材料势能衰减曲线,TNT势能衰减量大于TNT/C10H8共晶,这表明同种温度条件下,TNT次级化学反应的放热量大于TNT/C10H8共晶的放热量。
含能材料高温热分解过程中,势能的变化和系统内化学反应进程紧密关联。各温度条件下,两种含能材料高温热分解过程中系统势能随时间的演化分布表明,系统首先吸收热量,势能快速达到峰值。此阶段对应分子由平动模态转换为振动模态,同时伴随着初始化学反应[34],此阶段可视为“平衡-诱导期(tE-I)”[3],且为吸热反应阶段。随后,伴随着次级放热化学反应的进行,势能开始衰减并逐渐达到平衡,并且系统势能衰减曲线可用下述指数函数[3-5]进行拟合:

图2 TNT/C10H8共晶及TNT热分解势能演化曲线Fig.2 Evolution curves of potential energy of thermal decomposition for TNT/C10H8cocrystal and TNT supercell

式中:U(t)为t时刻系统势能(kcal/mol);U∞为势能渐进平衡值(kcal/mol);ΔUexo为次级反应放热量(kcal/mol);tE-I为平衡-诱导期(ps);τ为特征反应时间(ps,反应速率的倒数)。部分拟合曲线如图3黑线所示,拟合得到的各动力学以及热力学参数如表1.
拟合结果显示,随着温度的升高,含能材料高温热分解的“平衡-诱导期”和特征反应时间越短,也就是初级吸热反应,势能增加至峰值的时间以及次级放热反应达到平衡所需的时间越少。并且,同温度条件下,TNT高温热解的放热量均明显高于TNT/ C10H8共晶。
系统势能峰值为高温热分解过程中含能材料吸热和放热反应的分界点。为了比较TNT/C10H8共晶和TNT高温热分解吸热和放热反应的活化能,这里采用经典 Arrhenius方程,同时利用表1中 TNT/ C10H8共晶和TNT高温热解势能衰减曲线(TNT/C10H8共晶:3000 K、3 500 K、4 000 K、4 500 K;TNT:3 000 K、3 500 K、4 000 K)拟合得到“平衡-诱导期”、特征反应时间进行拟合:

式中:Ea为活化能;A为指前因子;R为理想气体常数。变换(2)式得到:lnτ=(Ea/RT)-lnA,可见lnτ 与1/T呈线性关系。拟合结果为TNT/C10H8共晶和TNT高温热解的吸热和放热反应阶段的活化能分别为

图3 两种含能材料高温热分解过程吸热和放热反应阶段的活化能Fig.3 Activation energy of endothermic and exothermic stages of thermal decomposition for cocrystal and TNT single crystal

表1 利用指数函数拟合得到的各动力学以及热力学参数Tab.1 Kinetic and thermodynamic parameters by exponential function fitting
吸热反应阶段系统势能快速增加,并且发生初始化学反应。对于TNT/C10H8共晶,晶胞内TNT分子上—NO2键的断裂,表明已经越过共晶热分解初级反应的能量势垒,并且此过程需要较小的时间尺度,因此共晶和TNT热分解初级吸热反应的活化能非常接近,且与Furman的模拟结果[34]基本一致。放热反应阶段主要发生次级反应并逐渐形成最终产物,TNT/C10H8共晶放热反应的活化能为TNT的2倍[34],此结果表明C10H8的加入抑制了共晶高温热分解次级放热反应的进行。TNT放热反应阶段活化能较Fruman模拟结果[34]高出35.6%.误差原因可能是由于当温度较高时,在25 ps的模拟时间内可以较好地描述初级和次级反应,而当温度相对较低时,热分解平衡则需要较长的时间,这将导致温度较低时的特征反应时间存在误差。另外,从结构上来说,共晶含能材料多为类三明治结构,相对惰性的C10H8分子的加入,降低了TNT的浓度,这将会使得相对TNT单晶来说,共晶发生次级化学反应的活化能增加,这与黑索今(RDX)高温热分解过程中发生次级化学反应的活化能随着RDX初始密度的降低而增加的结论基本一致[3]。
2.2反应物衰减速率比较
图4为TNT/C10H8共晶热分解过程中,超胞内TNT及C10H8分子数量衰减曲线比较。整体来说,TNT和C10H8分子均随着温度的升高,完全分解所需的时间越少。当温度为2 000 K时,共晶内TNT以及C10H8分子均部分“分解”,对比初始反应路径(见图5),共晶以及TNT吸收热量后可能形成大的分子团簇,至模拟后期,TNT分子脱落NO2使得TNT分子的数量进一步减少。当温度为2 500 K、3 000 K 和3 500 K时,共晶内TNT完全分解所需的时间分别为18.1 ps、10.2 ps和3.8 ps,而C10H8分子完全分解的时间均大于25 ps,并且当温度为4 000 K和4 500 K时,共晶内TNT分子较C10H8分子均提前完全分解。为了定量比较高温热分解过程中共晶内TNT和C10H8衰减速率以及C10H8对共晶内TNT衰减速率的影响。这里采用递减的指数函数对共晶内TNT和C10H8以及TNT单晶衰减曲线进行拟合[4]:

式中:C0为晶体中TNT和C10H8分子的初始数量(共晶内TNT和C10H8:32,TNT单晶:64);C(t)为t时刻TNT或C10H8分子的数量;k1为衰减速率(ps-1)。通过(3)式分别对共晶中TNT和C10H8分子以及TNT单晶的分解演化曲线进行拟合(如图4中黑色线所示)得到衰减速率k1如表2所示。
共晶内TNT和C10H8分子衰减速率均随着温度的升高而加快,并且TNT分子的衰减速率均高于C10H8.比较共晶内TNT分子和TNT单晶内分子数量的衰减速率,同温度条件下,TNT单晶的衰减速率高于共晶内TNT,因此,C10H8对共晶内TNT分子的热分解有抑制作用。
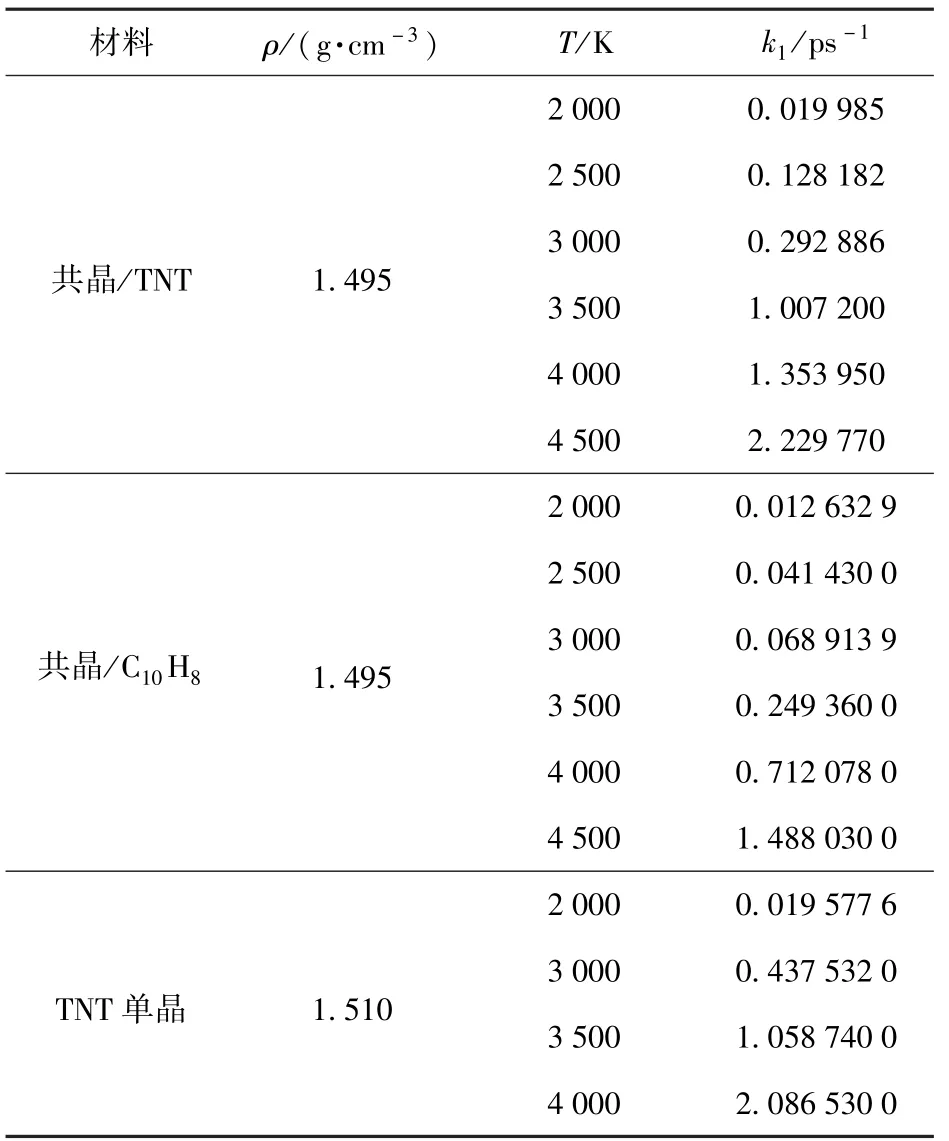
表2 反应物衰减速率比较Tab.2 Attenuation rates of reactants

图4 反应物分子数量衰减曲线及拟合Fig.4 Decay curves of reactant molecules and curve fitting
2.3初始反应路径
1)NO2的出现预示着硝基型含能材料高温热分解过程的能量势垒被克服[3]。气相硝基型含能材料热分解的活化能主要由NO2的脱落决定[34]。而固相硝基型含能材料在脱去NO2的同时还将发生一系列双分子链式反应。Cohen[38]利用可测性设计(DFT)方法分析了气相TNT高温热分解过程,并得出可能的3种初始反应路径:①C—NO2键断裂形成NO2(g)以及C7H5O4N2;② 甲基上C—H键断裂和邻位NO2上的O结合形成H2O和C7H3O5N3(DNAn);③TNT分子上—NO2异构形成O—NO,最后形成NO(g).并且当温度高于1 700 K时,气相TNT分解的主要初始反应路径为—NO2键断裂,其他的路径可以忽略。

图5 TNT/C10H8共晶和TNT单晶主要初级产物演化分布Fig.5 Distributions of main primary products for TNT/C10H8cocrystal and TNT single crystal
产物识别分析表明,NO2、NO和HONO为两种含能材料的主要初级产物。图5为TNT/C10H8共晶和TNT单晶高温热分解初始反应路径的比较。对于共晶,NO2、NO、HONO等初级产物主要来自于共晶内TNT分子。为了直观地比较共晶和单晶热分解的初始反应路径以及共晶内C10H8对TNT热分解的影响,纵坐标为各主要初级产物数量与初始TNT分子数量的比值(共晶:初级产物量/TNT分子数(32),TNT单晶:初级产物量/TNT分子数(64))。初级产物的演化分布显示两种含能材料在吸热阶段发生初始热分解,且初级产物不断累积,至峰值后,各产物逐渐地加入到次级反应中,进而形成最终的N2、H2O、CO2和 CO.NO2和 TNT—NO2,NO 和TNT—NO,HONO和TNT—HONO分布数量上的较大差异表明,并非TNT分子直接脱落形成各主要初级产物,而是 TNT-H/TNT+H与 C10H8分子(共晶)和TNT-H与TNT+H分子(TNT单晶)首先结合随后脱落形成初级产物。反应分子动力学模拟已经论证固相TNT高温热分解的反应路径为相邻的TNT分子间转移H原子,形成TNT-H和TNT+H,此过程需要克服的能量势垒为43 kcal/mol[34],小于单分子TNT直接脱落—NO2所需的活化能(61.7 kcal/mol)[38]。并且TNT+H将进一步分解形成HONO.对比共晶和TNT单晶内各主要初级产物的演化分布,同温度条件下,共晶热分解形成的初级产物量整体上小于TNT热分解形成的产物数量,主要原因是由于固相TNT热分解的初始路径为双分子反应机制,C10H8的加入,将限制TNT分子间的H原子转移,进而对初级产物的分布造成影响。热分解初期C10H8不会发生开环断裂,而是与共晶内TNT形成的各产物结合,使得初级产物的量进一步减少,这主要是由于C—C键离解能 (BDE)(100~150 kcal/mol),远远大于 C—N键(约 70 kcal/mol)、O—N(约50 kcal/mol)[38]。
2.4最终气相产物分析
液态TNT高温热分解最终气相产物为H2O、CO2、N2、H2、NH3、CO、OH及大的团簇[39]。本文模拟结果显示共晶以及固相TNT高温热分解的主要最终产物为CO、CO2、N2和H2O,其中TNT高温热分解的主要最终产物分布与Furman的模拟结果[34]一致。比较TNT爆轰温度条件下(T=3 500 K)热分解最终产物与密封真空环境下 TNT(密度为1.53 g/cm3)爆轰实验[40]测得的产物进行比较,产物类型一致,但前者最终产物的量整体上小于后者。究其原因,实验值为等熵膨胀后得到的气体产物量,而本文采用NVT系综,固定体积,且有限的模拟时间将共同造成最终产物的数量产生明显差异。
比较至模拟结束时刻,两种含能材料高温热分解形成的主要最终产物的数量,TNT单晶内,平均每个TNT分子产生的CO、CO2和N2的数量高于共晶内每个TNT分子产生的数量,而H2O的产量则相反。这主要是由于共晶内C10H8分子上C—H键断裂所需的能量为 C—H(约100 kcal/mol),小于C—C键断裂需要的能量(约100~150 kcal/mol)[38],因此,共晶高温热分解过程中,C10H8分子开环断裂前,C—H键断裂,脱落形成的H原子,将促进共晶内H2O的形成。另外,两种含能材料高温热分解最终产物(N2、H2O、CO和CO2)的演化分布同样满足指数函数特征[4]:
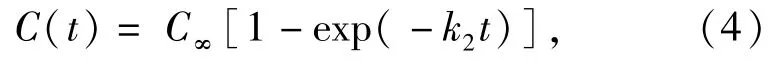
式中:C∞为至模拟结束时各主要产物分子数量的渐进分布值;k2为产物形成的速率常数(ps-1)。利用上述指数函数对两种含能材料热分解最终产物分布曲线进行拟合得到N2,H2O以及CO2的产生速率如表3所示(CO数量较少,未对其分布曲线拟合,拟合曲线如图6所示)。

表3 主要产物产生速率比较Tab.3 Comparison of production rates for main products

图6 主要最终产物演化分布及曲线拟合Fig.6 Time evolution curves of main final products and curve fitting
拟合结果显示,TNT/C10H8共晶及TNT单晶高温热分解最终产物的产生速率均随着温度的升高而加快。当温度为2000 K、2500 K时,至模拟结束时,TNT/C10H8共晶高温热解产物未出现N2和CO2.当温度为3 000 K~4 500 K,H2O分子的产生速率>N2>CO2.对于TNT单晶,H2O分子的产生速率>N2≈CO2.并且,各温度条件下共晶内H2O的产生速率均高于TNT单晶。此现象可用上述共晶内C10H8分子上C—H键断裂形成的H原子促进了H2O的产生速率加以解释。
3 结论
ReaxFF分子动力学方法已经在含能材料热分解、冲击起爆、爆轰稳定性等问题得到广泛应用,且较实验技术得到了更多的微观细节,但当前的研究对象多为常规含能材料。本文利用增加了长程修正项Elg的反应力场分析了TNT/C10H8共晶初始高温热分解情况,并通过TNT单晶高温热分解进行了对比分析。研究结果表明,同温度条件下,TNT单晶放热量高于TNT/C10H8共晶,并且利用经典反应速率方程拟合得到共晶高温热分解初级吸热和次级放热反应的活化能分别为35.7 kcal/mol和56.1 kcal/mol.共晶热分解初级吸热阶段的活化能和TNT单晶基本相同,这主要是由于共晶内TNT分子上—NO2键断裂,共晶的热分解能量势垒已经越过。次级放热反应的活化能明显高于TNT单晶,这主要是由于固相含能材料高温热分解的初始反应路径为双分子反应机制,C10H8的加入抑制了共晶热分解的放热反应。此结果可以与拟合得到的共晶和TNT单晶的反应放热以及共晶内TNT和TNT单晶内分子数量的衰减速率相互验证。共晶含能材料的热分解特征主要受晶体内材料自身物性以及二者相互作用机制的影响,并且具有类三明治结构的共晶与纯物理混合结构的含能材料热分解特征具有较明显的差别[41]。
产物识别分析显示,TNT/C10H8共晶和TNT共晶的初级反应产物为NO2、NO和HONO,并且通过NO2和TNT—NO2,NO和TNT—NO,HONO和TNT—HONO分布数量的比较论证了固相共晶和TNT单晶的初始反应路径为双分子反应机制。共晶和TNT单晶热分解的主要最终产物均为N2、H2O、CO2和CO.这主要是由于C10H8中C—C键断裂需要较高的能量,因此,C10H8热分解的初始反应路径为C—H键断裂,并且此过程将会促进共晶内H2O的形成。
当前,利用ReaxFF分子动力学通过施加Chapman-Jouget点温度模拟含能材料爆轰 Chapman-Jouget点后方的初始反应路径和最终产物,但相对爆轰来说,此过程并没有涉及压力条件,另外,还需要含能材料爆轰Chapman-Jouget点密度也是一个重要因素。因此,高温热分解模拟仅能对含能材料爆轰波内的反应情况提供参考。除此之外,利用ReaxFF分子动力学模拟缺乏自由边界条件,并且对系统体积加以固定,这将与实验结果造成差异,因此,未来需要建立真实的初始和边界条件,同时不断完善势函数以模拟含能材料爆轰波结构内的反应情况。
(References)
[1] Fickett W,Davis W C.Detonation:theory and experiment[M]. New York:Dover Publications,2000:44-51.
[2] van Duin A C T,Dasgupta S,Lorant F,et al.ReaxFF:a reactive force field for hydrocarbons[J].Journal of Physical Chemistry A,2001,105(41):9396-9409.
[3] Strachan A,Kober E M,van Duin A C T,et al.Thermal decomposition of RDX from reactive molecular dynamics[J].Journal of Chemical Physics,2005,122(5):054502.
[4] Rom N,Zybin S V,van Duin A C T,et al.Density-dependent liquid nitromethane decomposition:molecular dynamics simulations based on ReaxFF[J].Journal of Physical Chemistry A,2011,115(36):10181-10202.
[5] Zhang L Z,Zybin S V,van Duin A C T,et al.Carbon cluster formation during thermal decomposition of octahydro-1,3,5,7 tetranitro-1,3,5,7-tetrazocine and 1,3,5-triamino-2,4,6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations[J].JournalofPhysicalChemistryA,2009, 113(40):10619-10640.
[6] Shan T R,Wixom R R,Mattsson A E,et al.Atomistic simulation of orientation dependence in shock-induced initiation of pentaerythritol tetranitrate[J].Journal of Physical Chemistry B,2013,117(3):928-936.
[7] Wen Y S,Xue X G,Zhou X Q,et al.Twin induced sensitivity enhancement of HMX versus shock:a molecular reactive force field simulation[J].Journal of Physical Chemistry C,2013,117(46):24368-24374.
[8] Ge N N,Wei Y K,Song Z F,et al.Anisotropic responses and initial decomposition of condensed-phase β-HMX under shock loadings via molecular dynamics simulations in conjunction with multiscale shock technique[J].Journal of Physical Chemistry B,2014,118(29):8691-8699.
[9] Budzien J,Thompson A P,Zybin S V.Reactive molecular dynamics simulations of shock through a single crystal of pentaerythritol tetranitrate[J].Journal of Physical Chemistry B 2009,113(40):13142-13151.
[10] Cai Y,Zhao F P,An Q,et al.Shock response of single crystal and nanocrystalline pentaerythritol tetranitrate:Implications to hotspot formation in energetic materials[J].Journal of Chemical Physics,2013,139(16):164704.
[11] Zhang L Z,Zybin S V,Van Duin A C T,et al.Modeling high rate impact sensitivity of perfect RDX and HMX crystals by ReaxFF reactive dynamics[J].Journal of Energetic Materials,2010,28(1):92-127.
[12] He L,Sewell T D,Thompson D L.Molecular dynamics simulations of shock waves in oriented nitromethane single crystals[J]. Journal of Chemical Physics,2011,134(12):124506.
[13] An Q,Zybin S V,Goddard III W A,et al.Elucidation of the dynamics for hot-spot initiation at nonuniform interfaces of highly shocked materials[J].Physical Review B,2011,84(22):220101.
[14] An Q,Goddard III W A,Zybin S V,et al.Highly shocked polymer bonded explosives at a nonplanar interface:hot-spot formation leading to detonation[J].Journal of Physical Chemistry C,2013,117(50):26551-26561.
[15] Ken-Ichi N,Kalia R K,Aiichiro N,et al.Dynamic transition in the structure of an energetic crystal during chemical reactions at shock front prior to detonation[J].Physical Review Letters,2007,99(14):12243-12254.
[16] Ge N N,Wei Y K,Ji G F,et al.Initial decomposition of the condensed-phase β-HMX under shock waves:molecular dynamics simulations[J].Journal of Physical Chemistry B,2012,116(46):13696-13704.
[17] Landenberger K B.Cocrystallization of energetic materials[D]. Michigan:The University of Michigan.2013.
[18] Bolton O,Matzger J A.Improved stability and smart-material functionality realized in an energetic cocrystal[J].Angewandte Chemie International Edition,2011,50(38):8960-8963.
[19] Yang Z W,Li H Z,Huang H,et al.Preparation and performance of a HNIW/TNT cocrystal explosive[J].Propellants ExplosivesPyrotechnics,2013,38(4):495-510.
[20] Bolton O,Simke L R,Pagoria P F,et al.High power explosive with good sensitivity:a 2:1 cocrystal of CL-20:HMX[J].Crystal Growth&Design,2012,12(9):4311-4314.
[21] Yang Z W,Li H Z,Zhou X Q,et al.Characterization and properties of a novel energetic-energetic cocrystal explosive composed of HNIW and BTF crystal[J].Crystal Growth&Design,2012,12(11):5155-5158.
[22] Wei C X,Huang H,Duan X H,et al.Structures and properties prediction of HMX/TATB co-crystal[J].Propellants Explosives Pyrotechnics,2011,36(5):416-423.
[23] Landenberger K B,Matzger A J.Cocrystal engineering of a prototype energetic material:supramolecular chemistry of 2,4,6-trinitrotoluene[J].Crystal Growth&Design,2010,10(12):5341-5347.
[24] 刘海,李启楷,何远航.CL20-TNT共晶高温热解的ReaxFF/lg反应力场分子动力学模拟[J].物理学报,2013,62(20):208202. LIU Hai,LI Qi-kai,HE Yuan-hang.Pyrolysis of CL20-TNT cocrystal from ReaxFF/lg reactive molecular dynamics simulations[J].Acta Physica Sinica,2013,62(20):208202.(in Chinese)
[25] Guo D Z,An Q,Goddard W A,et al.Compressive shear reactive molecular dynamics studies indicating that cocrystals of TNT/ CL-20 decrease sensitivity[J].Journal of Physical Chemistry C,2014,118(51):30202-30208
[26] Sun T,Xiao J J,Liu Q,et al.Comparative study on structure,energetic and mechanical properties of aε-CL-20/HMX cocrystal and its composite with molecular dynamics simulation[J].Journal of Materials Chemistry A,2014,34(2):13898-13904.
[27] Groom C.The Cambridge Crystallographic data centre(CCDC)[DB/OL].[2015-8-22].http:∥www.ccdc.cam.ac.uk.
[28] Plimpton S.Fast parallel algorithms for short-range molecular dynamics[J].Journal of Computational Physics,1995,117(1):1-19.
[29] Plimpton S.LAMMPS Molecular dynamics simulator[CP/OL]. [2015-7-26].http:∥lammps.sandia.gov.
[30] Liu L C,Liu Y,Zybin S V,et al.ReaxFF-lg:correction of the ReaxFF reactive force field for London dispersion,with applications to the equations of state for energetic materials[J].Journal of Physical Chemistry A,2011,115(40):11016-11022.
[31] 宋华杰,周婷婷,黄风雷,等.低压长脉冲载荷下β-HMX单晶滑移系的微观物理化学响应[J].物理化学学报,2014,30(11):2024-2034. SONG Hua-jie,ZHOU Ting-ting,HUANG Feng-lei,et al.Microscopic physical and chemical responses of slip systems in the β-HMX single crystal under low pressure and long pulse loading [J].Acta Physico-Chimica Sinica,2014,30(11):2024-2034.(in Chinese)
[32] Zhou T T,Song H J,Liu Y,et al.Shock initiated thermal and chemical responses of HMX crystal from ReaxFF molecular dynamics simulation[J].Physical Chemistry Chemical Physics,2014,16(27):13914-13931.
[33] 刘海,李启楷,何远航.高速冲击压缩梯恩梯的分子动力学模拟[J].力学学报,2015,47(1):174-179. LIU Hai,LI Qi-kai,HE Yuan-hang.Molecular dynamics simulations of high velocity shock compressed TNT[J].Chinese Journal of Theoretical and Applied Mechani,2015,47(1):174-179.(in Chinese)
[34] Furman D,Kosloff R,Dubnikova F,et al.Decomposition of condensed phase energetic materials:interplay between uni-and bimolecular mechanisms[J].Journal of the American Chemical Society,2014,136(11):4192-4200.
[35] Robertson A J B.The decomposition,boiling and explosion of trinitrotoluene at high temperatures[J].Transactions of the Faraday Society,1948,44:977-983.
[36] Zinn J,Rogers R N.Thermal initiation of explosivers[J].Journal of Physical Chemistry 1962,66(12):2646-2653.
[37] Beckmann J W,Wilkes J S,McGuire R R.2,4,6-Trinitrotoluene thermal decomposition:kinetic parameters determined by the isothermal differential scanning calorimetry technique[J].Thermochimica Acta,1977,19(1):111-118.
[38] Cohen R,Zeiri Y,Wurzberg E,et al.Mechanism of thermal unimolecular decomposition of TNT(2,4,6-trinitrotoluene):a DFT study[J].Journal of Physical Chemistry A,2007,111(43):11074-11083.
[39] Rom N,Hirshberg B,Zeiri Y,et al.First-principles-based reaction kinetics for decomposition of hot,dense liquid TNT from ReaxFF multiscale reactive dynamics simulations[J].Journal of Physical Chemistry C,2013,117(41):21043-21054.
[40] Ornellas D L.Calorimetric determination of the heat and products of detonation for explosives,AD-A409329[R].Livermore,CA:Lawrence Livermore National Laboratory,1982.
[41] Guo D Z,An Q,Zybin S V,et al.The co-crystal of TNT/CL-20 leads to decreased sensitivity toward thermal decomposition from first principles based reactive molecular dynamics[J].Journal of Materials Chemistry A,2015,10(3):5409-5419.
Initial Thermal Decomposition of TNT/C10H8Cocrystal:Reactive Molecular Dynamics Simulations
LIU Hai,HE Yuan-hang
(State Key Laboratory of Explosion Science and Technology,Beijing Institute of Technology,Beijing 100081,China)
The discussion of shock initiation and detonation with ReaxFF reactive force field has centered nearly around conventional energetic materials.The comprehensive molecular dynamics of thermal decomposition of condensed phase TNT/C10H8cocrystal with the ReaxFF/lg potential function are studied,which adds a long range correction term to the total energy expression of original ReaxFF.The thermal decomposition of TNT single crystal is performed similarly to determine the main effect of C10H8on the overall thermal characteristics of cocrystal.Furthermore,the activation energies associated with endothermic and exothermic stages of cocrystal thermal decomposition are found to be 35.7 kcal/mol and 56.1 kcal/mol,respectively.Its activation energy at primary endothermic stage is compatible with that of TNT single crystal,but the activation energy at secondary exothermic stage is higher than the value calculated for TNT.In addition,the heat output calculated during the exothermic step of TNT/C10H8cocrystal is less than that of TNT at the same temperature.The decay rate of reactants by curve fitting shows thatC10H8inhibits the decomposition of TNT in cocrystal.NO2,NO and HONO are major primary decomposition products by the way of identification analysis.The distribution of NO2/TNT—NO2,NO/TNT—NO,HONO/TNT—HONOshows that the decomposition proceeds via bimolecular reaction.Main final products of cocrystal are N2,H2O,CO2,and CO,and the initial decomposition routes of C10H8is C—H bond dissociation as a result of C—C bond dissociation energy of C10H8in cocrystal is higher than C—H bond,and this process may promote the formation of H2O.
ordnance science and technology;cocrystal;pyrolysis;reaction dynamics;ReaxFF reactive force field
O64
A
1000-1093(2016)03-0414-10
10.3969/j.issn.1000-1093.2016.03.005
2015-04-03
刘海(1985—),男,博士研究生。E-mail:tristan_l@bit.edu.cn;何远航(1964—),男,教授,博士生导师。E-mail:heyuanhang@bit.edu.cn
