基于酵母表面展示技术的胸苷磷酸化酶全细胞催化剂的构建
2016-10-11王洁余磊杨东李婕王洪钟
王洁 余磊 杨东 李婕 王洪钟
(清华大学生命科学学院,北京 100084)
基于酵母表面展示技术的胸苷磷酸化酶全细胞催化剂的构建
王洁 余磊 杨东 李婕 王洪钟
(清华大学生命科学学院,北京 100084)
胸苷磷酸化酶(TP)在核苷类代谢通路中发挥重要作用,可催化生成多种核苷类似物。构建了TP的酵母表面展示系统作为全细胞催化剂。从大肠杆菌K12菌株中克隆编码TP的deoA基因,利用酵母表达质粒pKFS构建重组质粒,电击转化毕赤酵母GS115菌株。高拷贝阳性转化子经甲醇诱导96 h后,免疫荧光结果显示TP在酵母细胞表面成功展示。利用β-胸苷为底物,重组酵母细胞作为全细胞催化剂,经HPLC检测,结果表明展示在酵母表面的TP有催化活性,可以催化β-胸苷生成产物胸腺嘧啶。
胸苷磷酸化酶;全细胞催化;毕赤酵母;表面展示
近50年来,核苷类似物被广泛用于治疗病毒感染及肿瘤类疾病,其中已有超过9种抗代谢核苷类抗肿瘤药物和25种核苷类抗病毒药物经FDA批准上市[1]。核苷类似物是由人工改造天然核苷的糖基或碱基而合成的核苷类衍生物,由于和天然核苷结构相似,在生物体内会和天然核苷竞争,通过抑制相关聚合酶与核酸结合等方式干扰核酸的复制合成[2],达到治疗肿瘤类疾病或抑制病毒增殖的目的。
目前核苷类似物的工业合成方法主要是传统的化学合成法和生物转化法。化学合成反应步骤多,反应条件苛刻,产物纯度低[3];而生物转化法条件温和,成本低,应用较广泛。早期的生物转化法只能用来生产少数天然核苷;近年采用较多的生物转化法是酶法合成,通过将参与核苷代谢通路的酶重组转化到大肠杆菌中进行诱导表达,经过酶的纯化固定等方式来催化核苷类似物的合成[4]。本课题首次采用了一种新型生物转化方法,即开发于20世纪90年代的酵母表面展示法[5],来研究核苷类似物的生物酶催化。酵母表面展示法无需酶的纯化固定,便于遗传改造。它通过表达不同的锚定蛋白,可以将多种外源蛋白定位在酵母细胞壁上,利用全细胞作为催化剂进行生物催化过程,有可以反复多次利用、成本低的优点,已有报道成功利用酵母表面展示技术来展示a-半乳糖苷酶[5]、糖基转移酶[6]和脂肪酶[7]等酶,该方法也被广泛应用于生物催化剂、活疫苗、蛋白质文库筛选和癌症诊断等领域。
核苷类似物的生物催化法常用的酶包括N-脱氧核糖转移酶和核苷磷酸化酶。核苷磷酸化酶包括胸苷磷酸化酶(TP)、尿苷磷酸化酶(UP)和嘌呤核苷磷酸化酶(PNP),通过两步反应可逆地催化核苷(或脱氧核苷)生成核糖-1-磷酸和碱基。本实验所研究的TP(EC 2.4.2.4)最先由Schwartz于1978年在E. coli K12中克隆纯化[8]。哺乳动物来源的TP同时可作为血管生成因子,在肿瘤组织中有过表达现象[9]。本研究旨在通过构建TP的毕赤酵母表面展示系统,并作为全细胞催化剂,催化β-胸苷转化为胸腺嘧啶的反应。
1 材料与方法
1.1 材料
pKFS质粒和毕赤酵母GS115菌株由华南理工大学的林影教授赠送,E. coli K12菌株由本实验保存,E. coli DH5α和pMD19-T质粒购自TaKaRa公司。
限制性内切酶、T4连接酶和4×蛋白质SDS PAGE上样缓冲液(40 mmol/L Tris-HCl pH8.0,200 mmol/L DTT,4% SDS,40% Glycerol,0.032% Bromophenol Blue)均购自TaKaRa公司,PCR产物回收试剂盒、质粒小提试剂盒和DNA trans2K Plus Marker购自TransGen公司,酵母基因组DNA提取试剂盒购自康为世纪,鼠源抗flag单抗、HRP标记的羊抗鼠二抗和Alexa Fluor 488标记的羊抗鼠二抗均购自中杉金桥公司,胸腺嘧啶、β-胸苷(HPLC级)购自大连美仑公司,基因测序和引物合成由睿博兴科公司完成。
1.2 方法
1.2.1 重组质粒的构建 实验所用引物见表1。pKFS质粒由林影教授改造pPIC9K质粒得到,pKFS带有絮凝素Flo1蛋白的N端絮凝功能结构域(874aa,Flo1p short chain,缩写为FS)锚定系统[10]。为构建灵活度更高的FS-TP融合蛋白,本研究设计了一段(G4S)3蛋白linker[7,11],加到正向克隆引物deoA-pKFS-F上,随后是deoA基因的5'端序列,为保证融合蛋白的完整表达,序列的起始密码子被移除。为便于检测外源蛋白,在deoA基因下游加入了编码flag标签(氨基酸序列为DYKDDDDK)的核苷酸序列,在设计下游引物时去掉了deoA基因的3'端序列的终止密码子。
deoA基因PCR产物连到pMD19-T质粒上进行测序。利用Mlu I和Not I内切酶双酶切pMD19-T-deoA和pKFS质粒,将deoA基因插入Flo1基因的下游,所得重组质粒为pKFS-LA,重组质粒转化到E. coli DH5α中,在氨苄抗性的LB板上涂布培养,利用deoA-pKFS-F1、deoA-pKFS-R1引物进行菌落PCR来筛选阳性克隆。

表1 实验所用引物
1.2.2 重组质粒转化GS115和重组转化子的筛选 制备GS115酵母细胞的感受态,重组质粒经Sal I线性化后,利用电击仪(Bio-Rad)进行电击,转化到GS115酵母细胞。电转参数为1.5 kV,2 ms。转化子涂布到MD板培养6-7 d后,用MD液体培养基冲洗培养皿,洗脱下来的酵母悬液分别涂布到含1 mg/mL、2.5 mg/mL、5 mg/mL G418抗生素的MD平板上,培养5-6 d。挑取不同MD板的阳性单克隆扩大培养后提取基因组,进行基因组PCR鉴定。
1.2.3 胸苷磷酸化酶的诱导表达及免疫荧光检测 采用两步法诱导GS115表达外源蛋白。GS115野生菌株和经过验证的酵母转化子GS115-LA接种到BMGY培养基培养至OD为2-6,离心后分别用BMMY培养基重悬至OD为1左右,取2 mL细胞作为诱导0点4℃保存,之后加入1%的甲醇于20℃、200 r/min进行诱导,培养7 d,每隔24 h取样。
取培养96 h的1 mL GS115、GS115-LA细胞离心后,用1×PBS(pH7.0)洗3次,然后用含1 mg/mL BSA、2 mL鼠源抗flag单抗的1×PBS在室温下低速振荡孵育2 h,孵育结束后用1×PBS洗3次,用含1 mg/mL BSA、1 mL Alexa-Fluor 488标记的羊抗鼠二抗的1×PBS在黑暗环境下振荡孵育1 h。1×PBS洗涤3次后,用GE的DeltaVision高分辨率成像系统分别检测GS115和GS115-LA细胞的荧光。
1.2.4 Western blot检测胸苷磷酸化酶的诱导表达 准备1 mL样品,用破壁缓冲液室温处理30min,离心后加入20 mL 4×蛋白SDS-PAGE上样buffer和60 mL去离子水重悬,煮沸10 min后离心,取20 mL上清上样,进行SDS-PAGE电泳。电泳结束后,用Bio-Rad转膜仪将蛋白转移到PVDF膜上。转膜结束后,PVDF膜用5%奶粉的TBST封闭2 h,随后加入1 mL单抗(1∶500)室温孵育2 h,TBST洗3次;膜放入5 mL封闭液,加入1 mL HRP标记的羊抗鼠二抗室温振荡孵育1 h,结束后用TBST洗涤3次。用增强型HRP-DAB底物显色试剂盒(TIANGEN)对PVDF膜进行显色。
1.2.5 HPLC检测胸苷磷酸化酶的催化活性 配制不同浓度β-胸苷和胸腺嘧啶的标准品,分别制定标准曲线,以0.5 mL诱导96 h的GS115-LA细胞作为全细胞催化剂,GS115细胞作为对照,用50 mmol/L PBS(pH7.5)缓冲液配制含30 mmol/L胸苷的10mL反应体系,50℃振荡2 h,进行催化反应。反应样品20倍稀释后用Waters 600E高效液相色谱仪检测反应结果。检测条件为:紫外检测波长254 nm,柱温25℃,色谱柱 PLATISILTM ODS C18(5 mm,250×0.46 mm),流动相乙腈∶水=10∶90,进样量10 mL,流速1 mL/min。
2 结果
2.1 deoA基因重组质粒的构建及高拷贝转化子的筛选
编码TP的deoA 基因(EU275208.1)开放阅读框为1 323 bp,编码439个氨基酸,微生物来源的TP大小约47 kD。本实验利用了touch down PCR技术,设置退火温度由70℃梯度降落至60℃,将linker和flag序列分别加到deoA基因的上下游。
图1-A显示了约1 392 bp的降落PCR产物条带,测序结果证实重组质粒上E. coli K12来源的deoA基因序列和NCBI数据库已有序列一致,deoA基因上下游分别有linker和flag的核苷酸序列。
图1-B为不同浓度G418-MD板上的酵母转化子基因组以deoA-pKFS-F1、deoA-pKFS-R1为引物的PCR结果。为降低假阳性的影响,根据FS部分片段设计了FS-F、FS-R引物。以图1-B中的阳性克隆对应的基因组为模板,用pPIC9K通用引物5' AOX1、3'AOX1引物和FS-F、FS-R引物分别进行PCR,鉴定结果如图1-C、D。FS片段对应的基因序列为2 622 bp,FS-deoA基因序列约4 024 bp。PCR产物经测序,未出现移码突变。根据PCR结果,选出了拷贝数较高的5 mg/mL G418-MD板上的酵母转化子,进行后续诱导表达融合蛋白的实验。
2.2 免疫荧光检测酵母
取诱导96 h的酵母转化子和野生酵母做免疫荧光检测(FITC,激发波长488 nm),图2-A显示酵母转化子在AlexaFluor 488荧光下被标记为绿色,部分野生酵母细胞有微弱的自荧光。结果表明Flo1蛋白N端亚基可以作为表面展示的锚定蛋白,带有flag标签的TP被成功锚定在细胞壁上,呈不均匀的分布状态。
2.3 Western blot检测融合蛋白的诱导表达
将不同诱导时间点的酵母转化子破壁,提取细胞壁蛋白做Western blot。Western blot结果(图3)证实毕赤酵母在甲醇诱导的第3、4天是外源蛋白表达关键期,表达量相对较高。

图1 deoA序列的PCR扩增和阳性酵母转化子鉴定
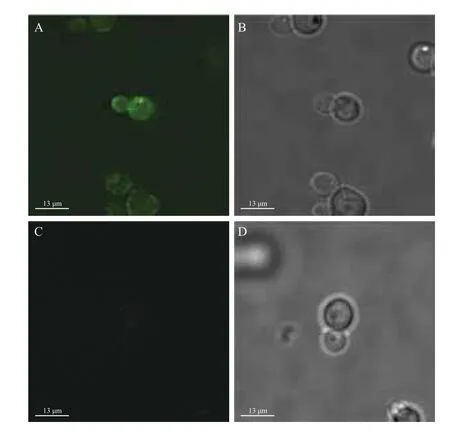
图2 酵母的免疫荧光检测结果
E. coli来源的TP为47 kD,FS-TP融合蛋白共有1 338 aa,理论分子量大小约为141 kD,而图3所检测到的蛋白条带约70 kD,与理论预测不太相符。这种情况在其它文献中也有报道[12,13],酵母细胞在翻译表达外源蛋白时,若出现连续两个或两个以上的碱性氨基酸相邻的结构,这一结构很容易被一些酵母本身表达的蛋白酶如Kex2切断,导致外源蛋白不完整。经过序列分析,融合蛋白存在一处“RKKR”结构,若经Kex2蛋白酶切割会得到大小为65 kD左右的融合蛋白,经过酵母的翻译后修饰如糖基化等过程,可能导致表观分子量呈现为70 kD左右;另一方面,毕赤酵母表达分泌蛋白的机制比较复杂,酵母自身分泌的胞外酶以及细胞裂解后释放的胞内酶,或破壁过程中的一些处理如超声等也许会导致外源蛋白降解。融合蛋白表观分子量变低的原因还需要进一步研究。
2.4 TP的催化活性检测
图4为反应体系稀释20倍后的HPLC检测结果,出现了产物胸腺嘧啶的色谱峰,以GS115原始菌株做对照的反应体系则未检测到产物峰的出现。计算得β-胸苷的转化率为7.5%。胸腺嘧啶和胸苷标准品的保留时间分别是7.079 min和9.692 min。分别在波长为264.1 nm和266.5 nm处有最大吸收峰,图4中的两个峰对应的波长分别与胸腺嘧啶和胸苷的标准品相一致,保留时间分别是7.761 min和10.444 min。

图3 融合蛋白的表达分析
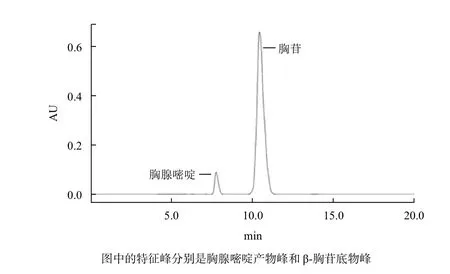
图4 HPLC检测图
3 讨论
本研究利用表面展示TP的酵母细胞作为全细胞催化剂,分别在30℃、40℃、50℃、60℃的反应温度下,以50 mmol/L pH7.5的磷酸盐缓冲液配制反应体系,以胸苷为底物进行催化反应,结果表明50℃是最优反应温度,在此温度下胸苷的转化率最高为7.5%。为探索不同pH值对反应的影响,制备了pH值为6.5、7.0、7.5、8.0的磷酸盐缓冲液,在50℃条件下进行了催化反应,结果表明pH7.5是最优反应pH值。但相比于表达TP的重组E. coli工程菌[14],毕赤酵母转化子的催化效率仍相对较低,文献利用E. coli BL21菌株共表达了TP和尿苷磷酸化酶(UP),在50℃时,将E. coli重组菌株与30 mmol/L尿嘧啶和60 mmol/L β-胸苷混合反应1 h,TP和UP共同催化生成了2-脱氧尿苷,反应的转化率可以达到61.6%。
Flo1蛋白的N端亚基可以非共价锚定TP的N端,使TP蛋白C端游离。微生物来源的TP为同源二聚体,每个亚基有一个磷酸根结合位点和一个脱氧核苷结合位点。当TP被锚定在细胞壁表面时,锚定蛋白可能影响到TP的空间构象和TP亚基之间的相互作用,继而阻碍TP对核苷类底物的催化,导致底物转化率较低。本实验室正在尝试利用另外的锚定蛋白和酵母展示系统来展示TP。
酵母表面展示TP还有待更深入的研究,需要选用更适合的锚定蛋白和展示系统,以进一步提高TP的活性和表达量。此外,利用毕赤酵母密码子偏好性[15],对deoA基因序列进行优化,并将毕赤酵母表面展示技术作为筛选手段进行定向进化[16],或许可以提高TP在酵母细胞内的表达量,进而筛选出催化能力更高的TP和稳定性高的展示系统,为开发高稳定性、高活性的表面展示TP全细胞催化剂奠定了基础。
本研究还尝试以5-氮杂胞苷为底物,但未能成功催化生成5-氮杂胞嘧啶,说明TP有一定的底物选择特异性,无法单独催化胞嘧啶核苷类衍生物的生成。有文献报道TP可以催化嘧啶类脱氧核苷酸或5-位取代的尿嘧啶类化合物,但脱氧胞苷类和6-C被甲基或酮基取代的核苷类化合物除外,另外TP也不能以尿嘧啶和胸腺嘧啶的氮杂类衍生物为底物[17,18]。
在催化合成核苷类衍生物的过程中,可尝试利用酵母表面展示系统同时展示两种核苷磷酸化酶,进行双酶反应[19],提高全细胞催化剂对核苷类化合物的底物适应性,来催化合成更多的非天然核苷类似物。
4 结论
本研究利用酵母表达质粒pKFS,首次构建了胸苷磷酸化酶的毕赤酵母表面展示系统。通过两步法的发酵方法,诱导酵母表达外源蛋白TP,确定了甲醇诱导的第3、4天是酵母表达外源蛋白TP的高峰期。并将展示TP的酵母细胞作为全细胞催化剂,以胸苷为底物催化合成了胸腺嘧啶。
[1] Jordheim LP, Durantel D, Zoulim F, et al. Advances in thedevelopment of nucleoside and nucleotide analogues for cancer and viral diseases[J]. Nat Rev Drug Discov, 2013, 12:447-464.
[2] Ewald B, Sampath D, Plunkett W. Nucleoside analogs:molecular mechanisms signaling cell death[J]. Oncogene, 2008, 27:6522-6537.
[3] Merino P. Chemical synthesis of nucleoside analogues[M]. New Jersey:John Wiley & Sons, Inc. , 2013.
[4] Ghaly AE, Dave D, Brooks MS, et al. Production of biodiesel by enzymatic transesterification:review[J]. Am J Biochem Biotechnol, 2010, 6:103-110.
[5] Pepper LR, Cho YK. A decade of yeast surface display technology:Where are we now?[J]Comb Chem High Throughput Screen,2008, 11(2):127-134.
[6] Abe H, Shimma Y, Jigami Y. In vitro oligosaccharide synthesis using intact yeast cells that display glycosyltransferases at the cell surface through cell wall-anchored protein Pir[J]. Glycobiology, 2003,13:87-95.
[7] Washida M, Takahashi S, Ueda M, et al. Spacer-mediated display of active lipase on the yeast cell surface[J]. Appl Microbiol Biotechnol, 2001, 56:681-686.
[8] Friedkin M, Roberts D. The enzymatic synthesis of nucleosides. I. Thymidine phosphorylase in mammalian tissue[J]. J Biol Chem,1954, 207:245-256.
[9] Akiyama S, Furukawa T, Sumizawa T, et al. The role of thymidine phosphorylase, an angiogenic enzyme, in tumor progression[J]. Cancer Sci, 2004, 95(11):851-857.
[10] Su GD, Huang DF, Han SY, et al. Display of Candida antarctica lipase B on Pichia pastoris and its application to flavor ester synthesis[J]. Appl Microbiol Biotechnol, 2010, 86(5):1493-1501.
[11] Chen X, Zaro JL, Shen WC. Fusion protein linkers:property, design and functionality[J]. Adv Drug Deliv Rev, 2013, 65(10):1357-1369.
[12] 韩双艳, 韩振林, 林影, 等. 高效絮凝素毕赤酵母表面展示系统的构建[J]. 生物化学与生物物理进展, 2010, 37(2):200-207.
[13] 张溪, 韩双艳, 苏国栋, 等. 外源脂肪酶在毕氏酵母表面展示及发酵过程分析[J]. 现代食品科技, 2010, 26(1):9-13.
[14] Xiong SL, Wang YB, Wang X, et al. Enzymatic synthesis of 2’-deoxyuridine by whole cell catalyst co-expressing uridine phosphorylase and thymidine phosphorylase through auto-induction system[J]. J Biosci Bioeng, 2014, 118(6):723-727.
[15] Lanza AM, KA Curran, LG Rey, et al. A condition-specific codon optimization approach for improved heterologous gene expression in Saccharomyces cerevisiae[J]. BMC Syst Biol, 2014, 8:33-43.
[16] Matsumoto T, Fukuda H, Ueda M, et al. Construction of yeast strains with high cell surface lipase activity by using novel display systems based on the Flo1p flocculation functional domain[J]. Appl Environ Microbiol, 2002, 68(9):4517-4522.
[17] Razzell WE, Casshyap P. Substrate specificity and induction of thymidine phosphorylase in Escherichia coli[J]. J Biol Chem,1964, 239(6):1789-1793.
[18] Serra I, Serra CD, Rocchietti S, et al. Stabilization of thymidine phosphorylase from Escherichia coli by immobilization and post immobilization techniques[J]. Enzyme Microb Technol, 2011,49(1):52-58.
[19] Xiong J, Zhang W, Su J, et al. Improved synthesis of 20-deoxyadenosine and 5-methyluridine by Escherichia coli using an autoinduction system[J]. World J Microbiol Biotechnol, 2012, 28:721-727.
(责任编辑李楠)
Construction of Pichia pastoris Surface Display Technology as Wholecell Biocatalyst for Thymidine Phosphorylase
WANG Jie YU Lei YANG Dong LI Jie WANG Hong-zhong
(School of Life Sciences,Tsinghua University,Beijing 100084)
Thymidine phosphorylase(TP)extensively involves in nucleoside metabolism and catalyzes the formation of many nucleoside analogs(NA). A yeast cell surface display system for TP was firstly constructed in this study as whole-cell biocatalyst. A deoA gene encoding TP was cloned from Escherichia coli K12 strain and inserted into the yeast expression vector pKFS. Recombinant vector was linearized and electro-transformed into Pichia pastoris GS115 cells. High copy transformant was induced by methanol for 96 h, and the results of immunofluorescence indicated that TP successfully displayed on the surface of P. pastoris. β-thymidine was used as substrate and recombinant GS115 cells as whole-cell biocatalyst, HPLC analysis demonstrated that TP on the surface of P. pastoris had catalytic activity, and catalyzed the production from β-thymidine to thymine.
thymidine phosphorylase;whole-cell catalysis;Pichia pastoris;surface display
10.13560/j.cnki.biotech.bull.1985.2016.01.032
2015-03-31
国家自然科学基金项目(21176138,21476124)
王洁,女,硕士,研究方向:生物转化及生物催化;E-mail:wangjie08beijing@163.com
王洪钟,男,博士,副教授,研究方向:生物转化及生物催化;E-mail:hzwang@mail.tsinghua.edu.cn
