酶催化多功能性研究进展
2016-09-27胡居吾付建平韩晓丹徐国良王慧宾熊伟
胡居吾,付建平,韩晓丹,徐国良,王慧宾,熊伟
(江西省科学院应用化学研究所,江西南昌 330096)
酶催化多功能性研究进展
胡居吾,付建平*,韩晓丹,徐国良,王慧宾,熊伟
(江西省科学院应用化学研究所,江西南昌 330096)
酶催化反应具有高选择性、条件温和和可再生等优点,是一种绿色环保的有机合成方法。近年来人们发现酶具有催化非天然底物发生天然反应或非天然反应的能力。这种独特的能力被定义为酶催化多功能性,近年来备受人们的关注,为有机合成开辟了新的途径。因此研究酶催化多功能性具有重要意义。本文叙述了酶的多功能性在催化aldol缩合,M annich反应,M ichael加成反应以及多米诺反应中的应用,展望了酶催化多功能性的研究趋势。
酶催化多功能性;合成;反应
酶一直以来被认为是一种高效、专一,对一定反应具有高度识别性的生物催化剂,且已被应用到有机化合物的合成中[1]。然而,近年来,越来越多的酶被发现可以催化除天然反应以外的一种甚至多种反应类型,这种特性被称为催化多功能性(Catalytic Promiscuity)。根据Hult和Berglund的报道[2]可以把酶催化多功能性分为3种类型。第一种,底物多样性,即一种酶可以催化非天然底物进行天然反应;第二种,反应条件多样性,即酶可以在不同于天然反应条件的催化环境中表现出催化活性,例如一些酶已经被证明可以在有机溶剂、各种pH和温度条件下催化有机反应。第三种,催化反应多样性,通过同一个活性位点或者不同活性部位亦或引入新的活性位点催化各种反应类型,这些反应在键的断裂、形成和机理上都与酶催化天然底物的反应不同[3]。一般说的酶催化的多功能性指的都是第三种类型,酶催化非天然底物的反应,这些反应中键的断裂、形成和机理都与酶催化天然底物反应不一样。当然,催化多样性都不可避免地会涉及到引发反应的条件、各种非天然底物和分子机理[4]。虽然酶催化多功能性这一概念相对来说比较新,但是这种多样性却是普遍存在于酶中。酶学的这块新领域还有几个重要的原理和实践应用。例如,酶催化多功能性是研究学进化的一个很好的出发点,已经在进化之间的相关性方面得到了应用[5]。研究催化多样性的机理和结构为开发已知酶新的催化功能提供了重要的切入点。这些具有催化多功能性的酶为有机合成提供了诸多新型催化剂。
1 酶催化反应媒介
长期以来,人们一直认为生物功能大分子如蛋白质、核酸等只能在水溶液中行使各自的生物功能,水溶液是这些大分子存在及其相互作用的天然介质。然而,工业上的许多反应都是在有机溶剂中进行的,很多化合物在水中不溶,水还会导致副反应的发生和腐蚀对水敏感的反应试剂等,以上这些原因大大限制了酶的工业用途。原则上,把酶的反应介质换成有机溶剂可以解决以上大部分问题。1984年,Zaks 和Klibanov首次报道了脂肪酶在有机溶剂中具有极高的热稳定性和较高的催化活性,这使酶在有机介质中催化作用的研究取得了突破性进展[6]。2001年,Klibanov更是系统地报道了酶在有机溶剂中的催化活性提高了,因为酶在有机介质中稳定性更高,还具有“分子记忆”(Molecular Memory)功能,而且有机溶剂对酶的空间和立体选择都有很大影响,有的时候甚至跟在水相中反应的结果相反。当然这不是说水在酶催化反应中的作用完全不重要,1995年,Giacomo Carrea报道了来自假丝酵母菌的脂肪酶催化动力学拆分,在有机溶剂中加入一定量的水,拆分效果达到最好[7]。加入一定量的水增加了酶的“柔性”,在最佳含水量时,蛋白质结构的动力学刚性(Kinetic Rigidity)和热力学稳定性(Thermodynamic Stability)之间达到最佳平衡点,酶才表现出最大活力(见图1)。
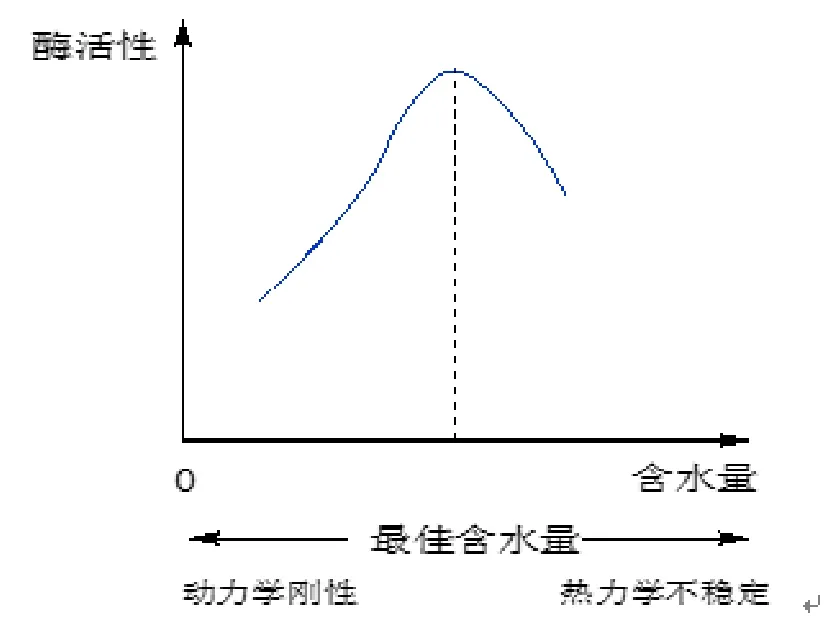
图1 “钢性”与“柔性”的动态平衡
有了以上的研究结果作为理论基础,就能利用酶来催化一些基本的反应类型,为构建C-C键的方法学开拓新的领域。事实上,近年来酶多功能性催化的aldol反应、Mannich反应、Micheal加成反应及多米诺反应都有了很大进展。本文重点介绍了这几类反应的研究进展。
2 酶多功能性催化的aldol反应
Aldol反应是构建C-C键的反应之一,并被广泛应用在许多复杂天然或非天然有机化合物的合成中[8, 9]。Aldol反应被发现以来,人们一直都在对其进行研究,寻找更好更有效的催化剂来不断改善它。最早用酶来催化aldol反应的人是Wong及其合作者,他们首次报道了用脱氧核糖-5-磷酸醛缩酶来催化aldol反应,他们的灵感出自醛缩酶的天然属性(见图2)[10]。2002年Berglund等首次报道了醛缩酶以外的酶催化aldol反应,他们利用生物工程改变脂肪酶B(来自假丝酵母菌)(Candida Antarctica lipase B,CAL-B)活性位点的氨基酸,结果得到了对aldol反应有催化作用的变异酶(见图3)[11]。2000年,List等和Barbas等分别报道了L-脯氨酸催化的不对称aldol反应,开辟了小分子催化有机反应的新领域,这种催化剂的设计思路来自于模拟生物酶的催化特点(见图4)[12]。这项研究成果大大激励了生物有机化学家们对酶催化aldol反应的兴趣。2008年,余孝其课题组报道了猪胰脂肪酶催化的丙酮和苯甲醛的直接不对称aldol反应,并取得了最高43.6%的ee值(见图5)[13]。当然,这个结果还可以有很大的提高空间。何延红课题组致力于这方面的研究,连续报道了木瓜凝乳蛋白酶催化的直接不对称aldol反应,获得了很好的反应结果(>99%收率,>99%的ee值)[14]。
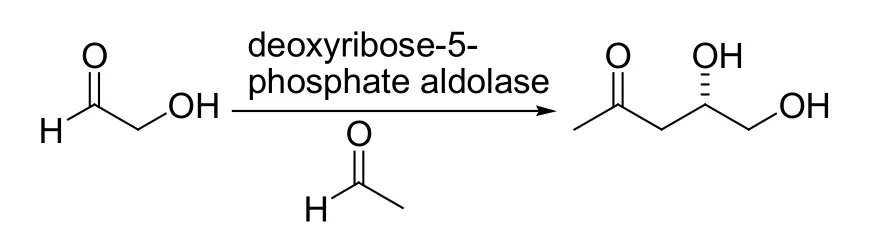
图2 首例酶催化的aldol反应
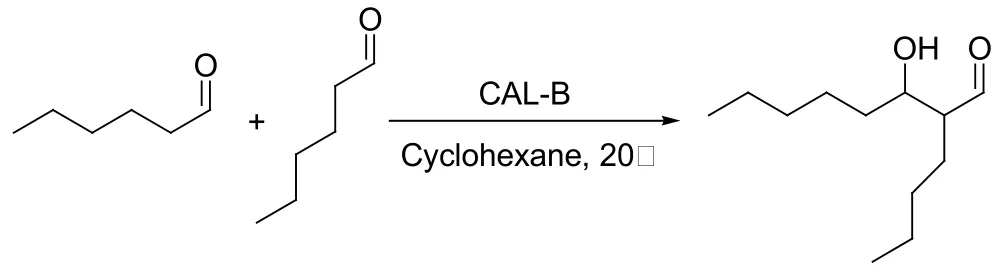
图3 脂肪酶B(来自假丝酵母菌)催化的己醛自身aldol反应

图4 L-脯氨酸催化的不对称aldol反应
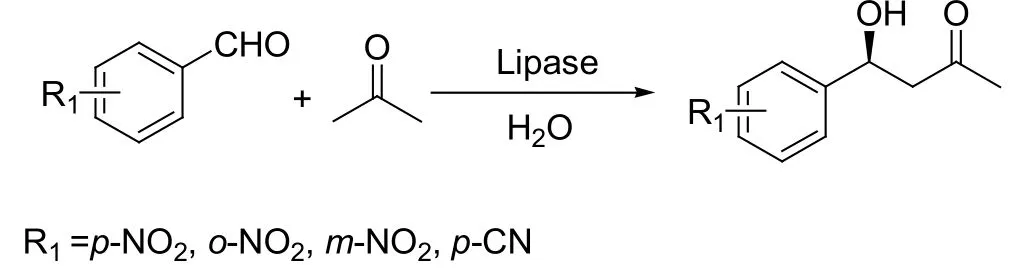
图5 首例酶催化的直接不对称aldol反应

图6 核酸酶催化的直接不对称aldol反应
3 酶多功能性催化的Mannich反应
Mannich反应是有机合成中形成C-C键和引入含氮化合物最常用最经济的方法之一,在各种药物和天然产物的合成中都有重要用途[15,16]。较理想的反应过程是三组分一锅法进行。然而,事实是,大部分的反应方法是两组份分步进行的,比如先生成亲电的亚胺或者亲核的烯醇和烯胺[17]。这些方法就降低了反应的原子经济性并且还增加了实验操作的复杂性。因此,Mannich反应一锅法作为符合绿色化学要求的实验策略,越来越受到人们的重视。用酶催化直接的Mannich反应更是最近几年才发展起来的新领域。2009年,余孝其和王娜等报道了脂肪酶催化的直接Mannich反应,来源于米黑毛霉菌和假丝酵母菌的脂肪酶催化活性较高,分别得到了72%和48%的产率,作者还发现反应体系中的含水量对酶的活性影响很大(见图7)[18]。2010年,他们又报道了来自皱褶念珠菌脂肪酶催化的直接Mannich反应,拓展了酶的多功能性和底物范围[19]。同年,章鹏飞课题组也报道了猪胰蛋白酶催化的直接Mannich反应,同样取得了很好的结果(见图8)[20]。

图7 脂肪酶催化的直接Mannich反应

图8 猪胰蛋白酶催化的直接Mannich反应
4 酶多功能性催化的Michael反应
4.1 C-C键的形成
Berglund和同事报道了第一个脂肪酶催化的1, 3-二羰基化合物和α, β-不饱和羰基化合物Michael反应的例子。他们将来自假丝酵母菌的脂肪酶B活性位点一个氨基酸取代掉,经过人工改造过的酶对Michael反应的催化能力比天然的酶要强的多(见图9)[21]。2009年,又报道了同样的变异酶催化丙烯酸甲酯与乙酰丙酮之间发生的Michael反应[22]。林贤福发现Zn2+依赖的酰基转移酶能催化1,3-二羰基化合物和甲基乙烯基酮的Michael反应[23,24]。Griengl和同事研究了有手型合成潜力的Michael反应(见图10)[25]。直到前不久,何延红课题组报道的固定化脂肪酶(Immobilized Lipase from Thermomyces Lanuginosu,Lipozyme TLIM)催化不对称Michael反应,才真正实现了酶催化Michael反应的不对称合成(见图11)[26,27]。
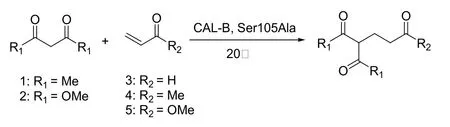
图9 CAL-B变体催化的1,3-二羰基化合物和α,β-不饱和羰基化合物Michae反应
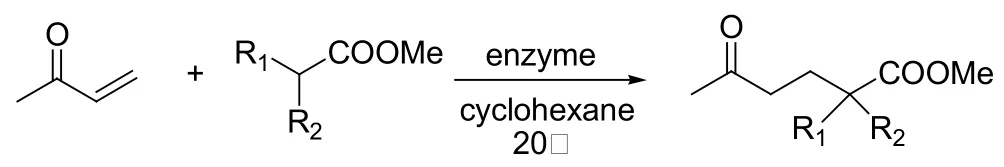
图10 具有手型合成潜力的Michael反应
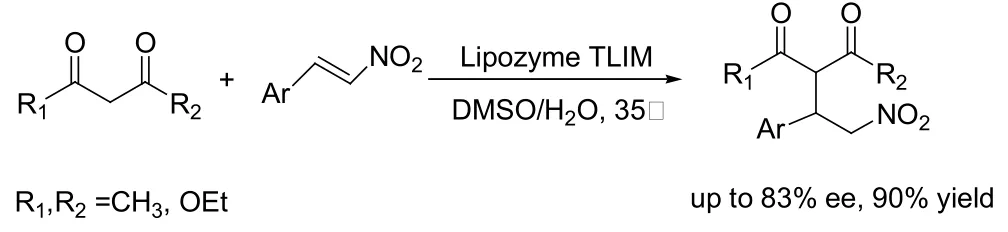
图11 固定化脂肪酶催化不对称Michael反应
4.2 C-N键的形成
2003年底,林贤福和同事首次报道了蛋白酶催化的氮杂Michael反应,他们分别用咪唑衍生物和嘧啶衍生物做亲核试剂进攻α, β-烯类化合物得到一系列的加成产物(见图12)[28]。Gotor和同事报道了CAL-B催化仲氨和丙烯氰的Michael加成反应(见图13)[29]。最近,Escalante和Castillo探究了溶剂对CAL-B催化的苯胺和巴豆酸甲酯发生Michael反应和氨解反应的影响,他们发现Michael加成更易在疏水性溶剂中进行,然而极性溶剂对酰胺的生成更有利(见图14)[30]。

图12 水解酶催化的咪唑,嘧啶,嘌呤参与的Michael反应
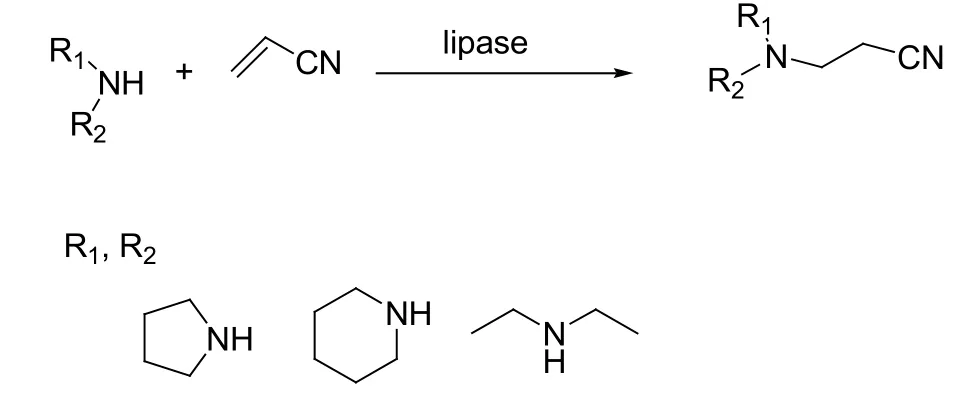
图13 脂肪酶催化的仲氨和丙烯氰之间的Michael反应
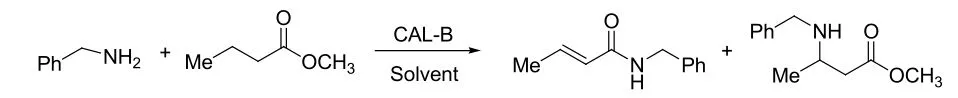
图14 CAL-B催化的苯胺和巴豆酸甲酯发生Michael反应
4.3 C-O,C-N键的形成
Kitazume等很早就把苯硫酚用作Michael加成的亲核试剂。他们报道了猪肝酯酶催化的苯硫酚与2-(三氟甲基)丙烯酸在水中的Michael加成反应[31]。在另外一篇报道中,他们用修饰过的酶催化三氟甲基取代的丙烯酸酯与苯硫酚的Michael反应,得到了有较好光学纯的产物(见图15)[32]。
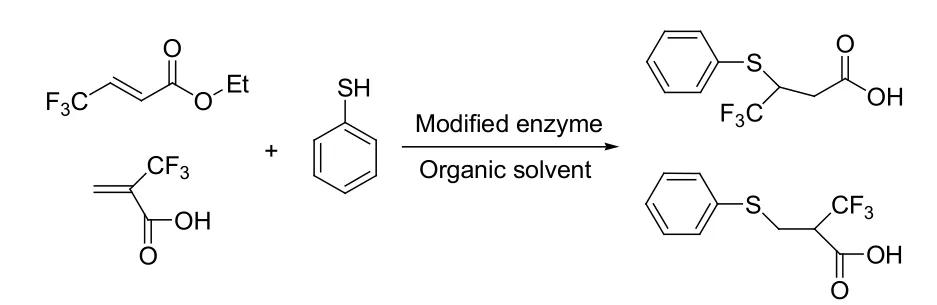
图15 苯硫酚与三氟取代α, β-不饱和化合物的Michael加成反应
由于氧原子相对差的亲核性,C-O键的形成较难,因此关于酶催化形成C-O键的Michael加成反应极少被报道。Whitman报道了细菌异构酶催化的3-E-氯和3-E-溴丙烯酸酯水合作用,生成的卤化醇不稳定,会分解为2-羰基丙酸盐,继续脱羧为乙醛(见图16)[33]。
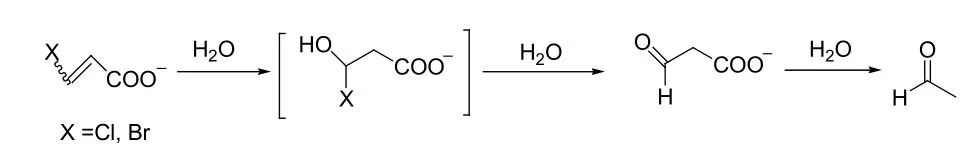
图16 细菌异构酶催化的3-E-卤丙烯酸酯水合作用
5 酶多功能性催化的多米诺反应
现代有机合成对化学工作者的要求不再只是拿到目标分子, 还要求化学家更加注重反应的效率和原子经济性。高效、高选择性、环境友好的绿色化学,已成为现代有机化学发展的趋势。关于酶催化的多米诺反应(也可称为串联反应)的报道也越来越多。早先的报道都是多种酶的串联反应,分别被应用于糖类衍生物[34]、头孢菌素[35]和核黄素等天然产物的合成[36]。但是,在这样的体系中每种酶要求的反应条件往往不一样,甚至有些酶对前一步反应有抑制作用[37]。单一酶催化的串联反应就能克服这些缺点。2004年,林贤福课题组报道了碱性蛋白酶(Alkaline Protease from Bacillus subtilis)催化的包含一个葡萄糖支链氮取代咪唑衍生物一锅法合成的例子,酶在吡啶的环境中成功催化了酰基化/Michael加成两步反应,得到了具有抗癌活性的产物(见图17)[38]。近一两年,关于单一酶催化的串联反应的报道更加密集。2011年,章鹏飞课题组就分别报道了猪胰脂肪酶(Lipase from Porcine pancreas,PPL)催化的螺氧化吲哚衍生物的合成[39]和四氢苯并吡喃衍生物的合成(见图18和图19)[40]。同年,林贤福等报道了CAL-B催化的著名Hantzsch反应,获得了一系列的1, 4-二氢吡啶衍生物[41]。何延红课题组亦探究了碱性蛋白酶(Alkaline Protease from Bacillus licheniformis,BLAP)催化的香豆素衍生物的合成,控制反应条件能实现Knoevenagel/分子内酯交换串联反应与Knoevenagel/半酮化串联反应之间的转化(见图20)[42]。以上的例子都清晰地给出了一个启示,未来酶多功能性催化反应的趋势就是酶催化多米诺反应。
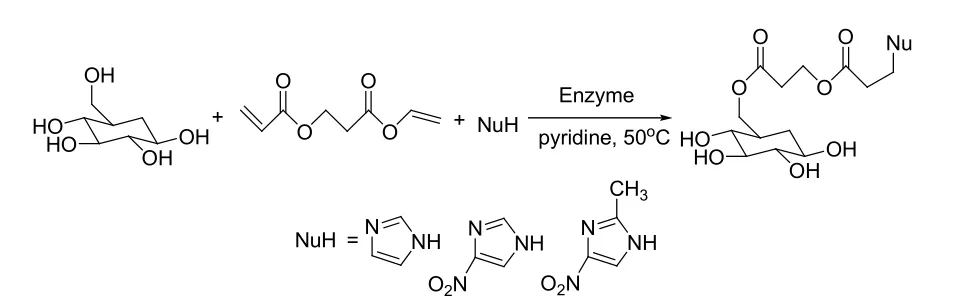
图17 碱性蛋白酶催化的氮取代咪唑衍生物的合成

图18 猪胰脂肪酶催化的Spirooxindole衍生物的一锅法合成

图19 猪胰脂肪酶催化的四氢可的索衍生物的一锅法合成

图20 碱性蛋白酶BLAP催化的香豆素衍生物的合成
6 结语
综上所述,酶促反应具有方法学上的重要意义,为有机合成提供了一种新型、有效且经济环保的途径。未来酶多功能性催化研究的主要方向应是酶催化多米诺反应以及不对称合成。
[1] Landwehr M, Hochrein L, Otey C R, et al. Enantioselective α-Hydroxylation of 2-Arylacetic Acid Derivatives and Buspirone Catalyzed by Engineered Cytochrome P450 BM-3[J] . J. Am. Chem .Soc, 2006, 128(18): 6058-6059.
[2] Hult K, Berglund P. Enzyme promiscuity: mechanism and applications[J]. Trends Biotechnol, 2007, 25(5): 231-238.
[3] Kazlauskas R J. Enhancing catalytic promiscuity for biocatalysis[J] . Curr. Opin. Chem. Biol, 2005, 9(2): 195-201.
[4] Nobeli I, Favia A D, Thornton J M. Protein promiscuity and its implications for biotechnology[J] . Nat. Biotechnol, 2009, 27(2): 157-167.
[5] Aharoni A, Gaidukov L, Khersonsky O, et al. The 'evolvability' of promiscuous protein functions[J] . Nat. Genet, 2005, 37(1): 73-76.
[6] Zaks A, Klibanov A M. Enzymatic catalysis in organic media at 100 degrees C[J] . Science, 1984, 224(4654): 1249-1253.
[7] Orrenius C, Norin T, Hult K, et al. The Candida antarctica lipase B catalysed kinetic resolution of seudenol in non-aqueous media of controlled water activity[J] . Tetrahedron Asymmetry, 1995, 6(12): 3023-3030.
[8] Trost B M, Fleming I, Heathcosk C H. Comprehensive Organic Synthesis[M] . Oxford: Pergamon, 1991(2): 409-439.
[9] Palomo C, Oiarbide M, Garcia J M. ChemInform Abstract: The Aldol Addition Reaction: An Old Transformation at Constant Rebirth[J] . Chemistry, 2002, 8(1): 36-44.
[10] Kimura T, Vassilev V P, Shen G H. Enzymatic Synthesis of β-Hydroxy-α-amino Acids Based on Recombinant d-and l-Threonine Aldolases[J] . J. Am. Chem. Soc, 1997, 119(49): 11734-11742.
[11] Branneby C, Carlqvist P, Magnusson A, et al. Carbon-Carbon Bonds by Hydrolytic Enzymes[J] . J. Am. Chem. Soc, 2003, 125(4): 874-875.
[12] List B, Lerner R A, Barbas C F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. Proline-Catalyzed Direct Asymmetric Aldol Reactions[J] . J. Am. Chem. Soc, 2000, 122(10): 2395-2396.
[13] Li C, Feng X W, Wang N, et al. Biocatalytic promiscuity: the first lipase-catalysed asymmetric aldol reaction[J] . Green Chem, 2008, 6(10): 616-618.
[14] Li H H, He Y H, Guan Z. Protease-catalyzed direct aldol reaction[J] . Catal. Commun, 2011, 12(7): 580-582.
[15] Muller R, Goesmann H, Waldmann H. N, N-Phthaloylamino Acids as Chiral Auxiliaries in Asymmetric Mannich-Type Reactions[J] . Angew. Chem. Int. Ed, 1999, 38(1): 184-187.
[16] Bohme H, Haake M. Advances in Organic Chemistry[M] . New York: Interscience, 1976: 107-223.
[17] Trost B M, Terrell L R. A Direct Catalytic Asymmetric Mannich-type Reaction to syn-Amino Alcohols[J] . J. Am. Chem. Soc, 2003, 125(2): 338-339.
[18] Li K, He T, Li C, et al. Lipase-catalysed direct Mannich reaction in water: utilization of biocatalytic promiscuity for C-C bond formation in a “one-pot” synthesis[J] . Green Chem, 2009, 11(6): 777-779.
[19] He T, Li K, Wu M Y, et al. Utilization of biocatalytic promiscuity for direct Mannich reaction[J] . J. Mol. Catal. B: Enzym, 2010, 67(s3-4): 189-194.
[20] Chai S J, Lai Y F, Zheng H, et al. A Novel Trypsin-Catalyzed Three-Component Mannich Reaction[J] . Helv. Chim. Acta, 2010, 93(11): 2231-2236.
[21] Svedendahl M, Hult K, Berglund P. Fast Carbon-Carbon Bond Formation by a Promiscuous Lipase[J] . J. Am. Chem. Soc, 2005, 127(51): 17988-17989.
[22] Svedendahl M, Jovanovic B, Fransson L, et al. Suppressed Native Hydrolytic Activity of a Lipase to Reveal Promiscuous Michael Addition Activity in Water[J] . Chem. Cat. Chem, 2009, 1(2): 252-258.
[23] Xu J M, Zhang F, Wu Q, et al. Hydrolase-catalyzed Michael addition of 1,3-dicarbonyl compounds to α, β-unsaturated compounds in organic solvent[J] . J. Mol. Catal. B: Enzym, 2007, 49(1): 50-54.
[24] Xu J M, Zhang F, Liu B K, et al. Promiscuous zinc-dependent acylasemediated carbon-carbon bond formation in organic media[J] . Chem. Commun, 2007, 20(20): 2078-2080.
[25] Strohmeier G A, Sovic T, Steinkellner G, et al. Investigation of lipase-catalyzed Michael-type carbon-carbon bond formations[J] . Tetrahedron, 2009, 65(s39-30): 5663-5668.
[26] Cai J F, Guan Z, He Y H. The lipase-catalyzed asymmetric C-C Michael addition[J] . J. Mol. Catal. B: Enzym, 2011, 68(s3-4): 240-244.
[27] Xu K L, Guan Z, He Y H. Acidic proteinase from Aspergillus usamii catalyzed Michael addition of ketones to nitroolefins[J] . J. Mol. Catal. B: Enzym, 2011, 71(71): 108-112.
[28] Lin X F, Cai Y, Wu Q, et al. Lipase and protease as novel catalysts for Michael addition in non-aqueous media[A] . Singapore International Chemical Conference 3, 2003, 80.
[29] Torre O, Alfonso I, Gotor V. Lipase catalysed Michael addition of secondary amines to acrylonitrile[J] . Chem. Commun, 2004, 15(47): 1724-1725.
[30] Priego J, Ortiz-Nava C, Carrillo-Morales M, et al. Solvent engineering: an effective tool to direct chemoselectivity in a lipase-catalyzed Michael addition[J] . Tetrahedron, 2009, 65(2): 536-539.
[31] Kitazume T, Ikeya T, Murata K. Folded conformation of 5,15-diarylporphyrins with anthraquinonyl and dimethylaminophenyl groups attached via a sulphonyloxy group[J] . J. Chem. Soc. Chem. Commun, 1986, 75(17): 1331-1333.
[32] Kitazume T, Murata K, Kokusho Y, et al. Enzymes active in organic media: Synthesis of optically active trifluoromethylated compounds via asymmetric addition reactions[J] . J. Fluor. Chem, 1988, 39(1): 75-86.
[33] Wang S C, Johnson W H, Whitman C P. The 4-Oxalocrotonate Tautomerase and YwhB-Catalyzed Hydration of 3E-Haloacrylates: Implications for the Evolution of New Enzymatic Activities[J] . J. Am. Chem. Soc, 2003, 125(47): 14282-14283.
[34] Miyazaki T, Sato H, Sakakibara T, et al. An Approach to the Precise Chemoenzymatic Synthesis of 13C-Labeled Sialyloligosaccharide on an Intact Glycoprotein: A Novel One-Pot [3-13C] -Labeling Method for Sialic Acid Analogues by Control of the Reversible Aldolase Reaction, Enzymatic Synthesis of [3-13C] -NeuAc-α-(2→3)-[U-13C] -Gal-β-(1→4)-GlcNAc-β-Sequence onto Glycoprotein, and Its Conformational Analysis by Developed NMR Techniques[J] . J. Am. Chem. Soc, 2000, 122(24): 5678-5694.
[35] Wegman M A, Van Langen L M, F Van Rantwijk R, et al. A two-step, one-pot enzymatic synthesis of cephalexin from D-phenylglycine nitrile[J] . Biotechnol. Bioeng, 2002, 79(3): 356-361.
[36] Romisch W, Eisenreich W, Richter G, et al. Rapid One-Pot Synthesis of Riboflavin Isotopomers[J] . J. Org. Chem, 2002, 67(67): 8890-8894.
[37] Schoevaart R, Van Rantwijk F, Sheldon R A. A Four-Step Enzymatic Cascade for the One-Pot Synthesis of Non-natural Carbohydrates from Glycerol[J] . J. Org. Chem, 2000, 65(21): 6940-6943.
[38] Yao S P, Lu D S, Wu Q, et al. A single-enzyme, two-step, one-pot synthesis of N-substituted imidazole derivatives containing a glucose branch via combined acylation/Michael addition reaction[J] . Chem. Commun, 2004, 36(17): 2006-2007.
[39] Chai S J, Lai Y F, Xu J C, et al. One-Pot Synthesis of Spirooxindole Derivatives Catalyzed by Lipase in the Presence of Water[J] . Adv. Synth. Catal, 2011, 353(2-3): 371-375.
[40] X u J C, Li W M, Zheng H, et al. One-pot synthesis of tetrahydrochromene derivatives catalyzed by lipase[J] . Tetrahedron 2011, 67(67): 9582-9587.
[41] Wang J L, Liu B K, Yin C, et al. Candida antarctica lipase B-catalyzed the unprecedented three-component Hantzsch-type reaction of aldehyde with acetamide and 1,3-dicarbonyl compounds in nonaqueous solvent[J] . Tetrahedron, 2011, 67(14): 2689-2692.
[42] Wang C H, Guan Z, He Y H, et al. Biocatalytic domino reaction: synthesis of 2H-1-benzopyran-2-one derivatives using alkaline protease from Bacillus licheniformis[J] . Green Chem, 2011, 8(51): 2048-2054.
Research Progress in Enzymatic Promiscuity
Hu Ju-wu, Fu Jian-ping*, Han Xiao-dan, Xu Guo-liang, Wang Hui-bin, Xiong Wei
(Institute of Applied Chemistry, Jiangxi Academy of Sciences, Jiangxi Nanchang 330029)
Enzymatic method is efficient and green tool for modern organic synthesis due to its high selectivity, mild conditions and potential use of inexpensive regenerable resources. It is very significative to profile the novel unnatural activities of existing enzymes systematically since it might lead to improvements in existing catalytic methods and provide novel synthesis pathways which are currently not available. This ability is defined as enzymatic promiscuity and has attracted much attention and expanded rapidly in recent years. Some elegant works of enzymatic promiscuity including aldol condensation, Mannich reaction, Michael additions, Markovnikov additions and Henry reactions have been reported in the last decades, and the future research direction is suggested in the paper.
Enzymatic promiscuity; Synthesis; Reaction
Q814
A
2096-0387(2016)01-0059-06
国家自然科学基金项目(项目编号31260400);江西省科技支撑计划项目(项目编号20141BBF60044)。
胡居吾(1977-),男,在读博士,副研究员,研究方向:天然产物化学。
付建平(1987-),男,硕士研究生,助理研究员,研究方向:天然产物化学。
