索非布韦关键中间体的合成工艺研究
2016-09-26王志刚郑和奇王治国
王志刚,郑和奇,余 微,王治国*
(1湖北理工学院 化学与化工学院,湖北 黄石 435003;2湖北理工学院 矿区环境污染控制与修复湖北省重点实验室,湖北 黄石 435003)
索非布韦关键中间体的合成工艺研究
王志刚1,2,郑和奇1,2,余微1,2,王治国1,2*
(1湖北理工学院 化学与化工学院,湖北 黄石 435003;2湖北理工学院 矿区环境污染控制与修复湖北省重点实验室,湖北 黄石 435003)
索非布韦是全球首个全口服抗丙型肝炎病毒药物。以2-溴代丙酸乙酯为起始原料,经过膦叶立德、Wttig、氧化、成环、氟化和官能团保护6步反应制备了索非布韦关键中间体(2R)-2-脱氧-2-氟-2-甲基-D-赤式戊糖酸γ-内酯-3,5-二苯甲酸酯,并对合成工艺进行了改进。改进后的工艺降低了成本,提高了反应器的容量,同时还提高了反应的收率,总收率由文献最高的29.0%提高到35.4%,具有一定的工业应用价值。
索非布韦;中间体;(2R)-2-脱氧-2-氟-2-甲基-D-赤式戊糖酸γ-内酯-3,5-二苯甲酸酯;合成工艺
0 引言
索非布韦(Sofosbuvir)化学名为 (S)-2-{(S)-{{(2R,3R,4R,5R)-5-[2,4-二氧代-3,4-二氢嘧啶-1(2H)-基]-4-氟-3-羟基-4-甲基四氢呋喃-2-基}甲氧基}(苯氧基)磷酰基氨基}丙酸异丙酯,其结构如图1所示,CAS登记号:1190307-88-0,商品名为Sovaldi,于2013年12月6日被美国FDA批准上市,是由Gilead Sciences 公司研发的核苷类NS5B聚合酶抑制剂,主要用于慢性丙型肝炎病毒(HCV)感染的治疗[1]。对于HCV基因1型(HCV GT1)患者,用干扰素治疗时间可缩短为12周,HCV基因2、3型患者在不联合聚乙二醇干扰素的情况下疗效依然非常显著, 索非布韦联合利巴韦林治疗基因2、3型患者的临床治愈率可达97%[2]。
索非布韦的合成一般是将 (2R)-2-脱氧-2-氟-2-甲基-D-赤式戊糖酸γ-内酯 3,5-二苯甲酸酯(化合物1)的羰基还原为羟基,再把羟基转化为卤素或乙酸酯的结构,与胞嘧啶衍生物反应形成糖苷,再与苯氧基磷酰胺基丙酸异丙酯的磷酰氯或磷酸酯发生磷酰化反应得到。因此化合物1是合成索菲布韦的关键中间体,对该化合物的合成工艺研究非常必要。
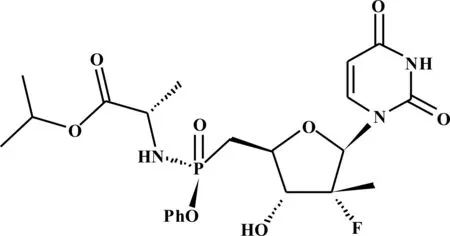
图1 索非布韦的结构
目前化合物1或其还原产物的合成方法按起始原料分类,主要有以下4种:①Clark等[3]采用D-木糖为原料,经过7步制得五元糖环的骨架,再经氧化,亲核加成,氟化等5步反应得到化合物1的还原产物,该路线合成步骤太长,总收率很低(2.4%)。②Mayers[4]采用六元内酯环为原料,先酯化,再引入氟原子,最后在酸性条件下缩环得到五元环,再羟基保护得到化合物1,该路线原料不易得到,并且引入氟原子存在消除反应的副反应,总收率很低(16.4%)。③Cedilote等[5]以(S)-2,2-二甲基-1,3-二氧戊环-4-甲醛为原料(化合物3,见图2),直接与2-氟丙酸酯或酰胺发生羟醛缩合反应得到对映异构体,分离后再关环得到化合物1。这条路线步骤很短,但对映异构体数量多,分离难度很大,收率较低(18.1%~23.0%)。Chen[5]采用手性底物价格比较昂贵。Wang[6]用2-溴丙酸酯发生Reformatsky反应,再用二乙胺基三氟化硫(DAST)引入氟原子,甲基锂加成,保护,关环,再保护,整条反应路线太长,条件苛刻,收率较低(19.5%)。④Axt等[7-9]将化合物3 与2-(三苯基膦烯)丙酸乙酯(化合物2)发生Wittig反应得到化合物4,经高锰酸钾氧化得到顺式邻二醇5,与二氯亚砜反应成环,次氯酸钠进一步氧化得到环砜6,与四乙基氟化铵(TEAF)作用开环,引入氟原子,再用浓盐酸环化得到中间体7,苯甲酰基保护得到关键中间体1,收率适中(24.5%~29.0%)。
第4种合成路线操作简便、收率较高,比较适用于工业化,本研究对文献[7-9]报道的方法进行了改进和优化,合成路线如图2所示。

图2 化合物1的合成路线
1 实验部分
1.1仪器和试剂
1)仪器:SGW X-5数字熔点测定仪(上海精密科学仪器有限公司);Varian Mercury plus 400核磁共振波谱仪(CDCl3为溶剂,TMS为内标)。
2)试剂:2-溴代丙酸乙酯(AR,成都格雷西亚化学技术有限公司);三苯基膦(AR,国药集团化学试剂有限公司);(S)-2,2-二甲基-1,3-二氧戊环-4-甲醛(AR,上海德默医药科技有限公司);四乙基氟化铵水合物(AR,萨恩化学技术上海有限公司);2,2-二甲氧基丙烷(AR,萨恩化学技术上海有限公司);其他试剂均为分析纯。
1.2实验部分
1.2.12-三苯基膦乙烯基丙酸乙酯(化合物2)的合成
将三苯基膦(104.8 g,0.20 mol)溶于甲苯(400 mL)中,分别加入水(300 mL)、2-溴代丙酸乙酯(86.4 g,0.48 mol),于65 ℃温度下搅拌30 h,再加入水(400 mL),分液,水层用甲苯(150 mL×3)洗涤,合并水相,室温下缓慢滴加5%的氢氧化钠溶液(352.0 g,0.44 mol),有黄色固体析出,滴加完毕后继续搅拌反应1 h,过滤,滤饼用水洗涤,减压干燥得到化合物2(123.2 g,85.1%),mp 158~160 ℃ (文献值[9]∶158~160 ℃,83.7%),1H NMR (400 MHz,CDCl3)δ∶7.70~7.40(m,15 H),4.12(q,J=7.2 Hz,2 H),1.94(s,3H),1.35(t,J=7.2 Hz,3H)。
1.2.2(2E)-3-[(4S)-2,2-二甲基-1,3-二氧戊环-4-基]-2-甲基-2-丙烯酸乙酯 (化合物4)的合成
将化合物2(100.8 g,0.30 mol)溶于无水二氯甲烷(500 mL)中,冷却至-20 ℃,将化合物3(32.4 g,0.36 mol)溶于无水二氯甲烷(200 mL)中,并缓慢滴加至化合物2的溶液中,恢复至室温,继续反应20 h。减压除去溶剂,残渣悬浮于甲基叔丁基醚(500 mL)中,过滤,固体用甲基叔丁基醚(200 mL×3)洗涤,合并滤液,减压除去溶剂得到浅黄色油状物(60.0 g,93.4%),不用纯化可直接进行下一步反应。1H NMR (400 MHz,CDCl3) δ:6.38(d,J=6.8 Hz,1 H),4.85(dd,J=6.8 Hz,7.6 Hz,1 H), 4.25~4.10(m,3 H),3.63(t,J=7.6 Hz,1 H),1.89(s,3 H),1.45(s,3 H),1.41(s,3 H),1.29(t,J=7.2 Hz,3 H)。
1.2.32-C-甲基-4,5-O-(1-甲基乙烯基)-D-阿拉伯糖酸乙酯 (化合物5)的合成
于0 ℃温度下,在1 L圆底烧瓶中分别加入化合物3(42.8 g,0.20 mol)、无水丙酮(250 mL)、乙二醇(40 mL)、碳酸氢钠(50.0 g)和水(50 mL),分批加入高锰酸钾(31.6 g,0.22 mol),保持反应温度在0 ℃下,继续搅拌2 h,加饱和亚硫酸氢钠(20 mL)溶液使反应猝灭,继续搅拌30 min,过滤除去二氧化锰固体,固体用乙酸乙酯(500 mL)洗涤,滤液用乙酸乙酯(200 mL×3)萃取,合并有机相,无水硫酸镁干燥,过滤,减压蒸出溶剂得到浅黄色固体。正己烷重结晶得到白色固体(36.4 g,73.4 %),mp 75~76 ℃ (文献值[7]:75.0~75.5 ℃),1H NMR(400 MHz,CDCl3) δ∶4.28(q,J=7.2 Hz,2 H),4.23~4.17(m,1 H),4.09~4.00(m,2 H), 3.61(d,J=6.8 Hz,1 H),3.61(s,1 H),2.49(bs,1 H)1.41(s,3 H),1.36(s,3 H),1.33(s,3 H),1.29(t,J=7.2 Hz,3 H)。
1.2.4(3R,4R,5R)-3-氟-4-羟基-5-(羟基甲基)-3-甲基四氢呋喃-2-酮(化合物7)的合成
于0 ℃温度下,在1 L圆底三口烧瓶中加入化合物5(37.2 g,0.15 mol),无水二氯甲烷(400 mL),三乙胺(62.4 mL,0.45 mol),缓慢滴加二氯亚砜(22.5 mL,0.3 mol),滴加完后继续搅拌1 h,再加入二氯甲烷(200 mL),反应液依次用冷水(100 mL×3)和饱和食盐水洗涤(100 mL×3),浓缩有机层后加入乙腈(300 mL),在0 ℃温度下,分别加入2,2,6,6-四甲基哌啶氧化物0.15 g、10%次氯酸钠溶液(300 mL),搅拌20 min,恢复至室温反应2 h,取有机层,干燥过滤,减压浓缩,用二氯甲烷(50 mL×2)共蒸发,得到无色液体36.0 g。未进一步纯化直接进行下一步反应。
将化合物6粗品(36.0 g)溶解在无水1,4-二氧六环(500 mL)中,加入四乙基氟化铵水合物(35.7 g,0.19 mol),在100 ℃下反应1 h。冷却至室温,分别加入2,2-二甲氧基丙烷(300 mL)、浓HCl(30 mL),继续搅拌反应3 h,加乙酸乙酯(300 mL),依次用饱和的NaHCO3(100 mL×3)和饱和食盐水(100 mL×3)洗涤,得有机层,干燥过滤,减压抽滤得到半固体化合物。溶于乙醇(300 mL)和浓盐酸(7.5 mL)中,40 ℃温度下搅拌10 h,蒸发浓缩,过滤干燥,得到白色固体,即化合物7(15.0 g,61.0%)。mp 142~144 ℃ (文献值[8]:142~143 ℃),1HNMR(400 MHz,CDCl3) δ∶4.56(m,1 H),4.12(dd,J=7.2 Hz,21.5 Hz,1 H), 3.95(dd,J=13.3 Hz,1.5 Hz,1 H),3.70(dd,J=13.3 Hz,3.3 Hz,1 H), 3.15(br,1 H),2.24(br,1H),1.55(d,J=22.4 Hz,3 H)。
1.2.5(2R)-2-脱氧-2-氟-2-甲基-D-赤式戊糖酸 γ-内酯 3,5-二苯甲酸酯 (化合物1)的合成
将化合物7(16.4 g,0.1 mol)溶于无水的四氢呋喃(200 mL)中,分别加入4-N,N-二甲基氨基吡啶(5.0 g,0.04 mol)、三乙胺(22.7 g,0.23 mol), 冰水浴冷却至0 ℃左右,缓慢滴加苯甲酰氯(42.0 g,0.3 mol), 滴加完毕室温继续反应4 h, TLC 显示原料消耗完毕,加水(100 mL)继续搅拌30 min,分液,水相用乙酸乙酯(100 mL×2)反萃取,合并有机相,无水硫酸镁干燥,过滤后减压除去溶剂,加入异丙醇(100 mL)在50 ℃温度下溶解,逐渐冷却至0 ℃,有白色固体析出,过滤,固体用冷的异丙醇洗涤,干燥,得到白色固体,即化合物1(38.5 g,93.0%)。mp 136~138 ℃ (文献值[7]:137.2~137.8 ℃),1HNMR (400 MHz,CDCl3)δ∶8.10~8.00(m,4 H),7.65~7.40(m,6 H),5.52(dd,J=17.8 Hz,5.8 Hz,1 H),5.10~5.00(m,1 H),4.77(dd,J=12.8 Hz,3.8 Hz,1 H),4.60(dd,J=12.8 Hz,5.2 Hz,1 H),1.69(d,J=22.4 Hz,3 H)。
2 结果与讨论
在化合物2的合成中,文献[9]加入了少量的碘化钾,由于碘化钾的价格较高,并且溴卤代烃的活性不是特别差,本研究不加碘化钾,适当升高反应的温度,延长反应时间,同时把α-溴代丙酸乙酯与三苯基磷的摩尔比由文献的1.5∶1降到1.2∶1,膦叶立德的收率(85.1%)比文献(83.7%)略有提高。
化合物4的合成是最难工业化的一步,烯烃一般采用稀冷的高猛酸钾氧化,文献中反应物和溶剂一般采用的比例为1∶15,本研究在反应中加入乙二醇避免产物的氧化,同时加入少量碳酸氢钠和水,使高锰酸钾在碱性中更好氧化,在保证反应温度的情况下,溶剂丙酮的用量可以减少到1∶5,大大提高了反应器的容量,反应收率也得到提升(由文献65.7%提高到73.4%)。
化合物6的合成分为2步,第1步是酯化,第2步是开环引入氟原子,分别采用三乙胺三氟化氢(NEt.3HF)、氟化铵、氟化氢铵、四乙基氟化铵不同的氟离子源进行反应,实验结果表明四乙基氟化铵的收率最高,其原因可能是氟离子更容易解离发生亲核开环反应。
化合物7的合成[7]一般是在无水吡啶中反应完后,加入水析出产品,收率很低(70.0%),本研究采用三乙胺为碱,在异丙醇中重结晶,收率可提高到93.0%。
3 结论
索非布韦是全球首个全口服抗丙型肝炎病毒药物,通过抑制NS5B聚合酶有效抑制HCV的复制从而达到治疗效果。本研究参考相关文献,研究了关键中间体化合物1的合成工艺条件,该工艺降低了成本,提高了反应器的容量,还提高了反应的收率,总收率由29.0%提高到35.4%,具有一定的工业应用价值。
[1]US FDA.FDA approval of Sovaldi(sofosbuvir)tablets for the treatment of chronic hepatitis[EB/OL].(2013-12-06)[2015-08-16].http://www.fda.gov/forconsumers/byaudience/forpatientadvocates/ucm377920.htm.
[2]Lawitz E,Mangia A,Wyles D,et al.Sofosbuvir for previously untreated chronic hepatitis C infection[J].New England Journal of Medicine,2013,368(20):1878-1887.
[3]Clark JL,Mason JC,Hobbs AJ,et al.Synthesis of 2-deoxy-2-fluoro-2-C-methyl-D-ribofuranoses[J].Cheminform,2006,38(4):461-470.
[4]Mayers BA,Moussa A.Process for preparing a synthetic intermediate for preparation of branched nucleosides[P].WO 2007/075876.2006-12-22.
[5]Chen R,Zhao J.Process for the preparation of a fluorolacton derivative[P].WO 2014/108525.2014-01-13.
[6]Chun B,Wang P.Preparation of 2′-fluoro-2′-alkyl-substituted or other optionally substituted ribofuranosyl pyrimidines and purines and their derivatives[P].WO2006/031725.2006-03-23.
[7]Wang P,Chun BK,Rachakonda S,et al.An efficient and diastereoselective synthesis of PSI-6130:a clinically efficacious inhibitor of HCV NS5B polymerase[J].Journal of Organic Chemistry,2009,74(17):6819-6824.
[8]Ishii A,Nagura H,Tsuruta H.Method for producing (2R)-2-fluoro-2-C-methyl-D-ribono-γ-lactone precursor[P].US 2013/0072699.2013-03-21.
[9]Kawada Y,Tanaka M.Efficient preparation of phosphorane compounds from halides and triarylphosphines via non-isolated phosphonium intermediates[P].JP 2008266160.2208-11-06.
(责任编辑高嵩)
Study on Synthesis Process of Key Intermediate of Sofosbuvir
Wang Zhigang1,2,Zheng Heqi1,2,Yu Wei1,2,Wang Zhiguo1,2*
(1School of Chemistry and Chemical Engineering,Hubei Polytechnic University,Huangshi Hubei 435003;2Hubei Key Laboratory of Mine Environmental Pollution Control and Remediation,Hubei Polytechnic University,Huangshi Hubei 435003)
Sofosbuvir is the first full oral anti hepatitis C virus drugs in the world.Using 2-bromo ethyl propionate as the starting material,after phosphine ylide,Wttig, oxidation, closed ring,fluoride and functional group protection six step reactions for preparing the key intermediate of Sofosbuvir,((2R,3R,4R)-3-(Benzoyloxy)-4-fluoro-4-methyl-5-oxotetra-hydrofuran-2-yl)methyl benzoate,and the synthesis process was improved.The improved process can reduce the cost,improve the capacity of the reactor,and increase the yield of the reaction.The total yield increases from 29% to 35.4%,The improved process has a certain value for industrial applications.
Sofosbuvir;intermediate;((2R,3R,4R)-3-(Benzoyloxy)-4-fluoro-4-methyl-5-oxotetra-hydrofuran-2-yl)methyl benzoate;synthesis process
2016-05-21
湖北理工学院实验室开放基金项目。
王志刚,讲师,硕士。
王治国,副教授,博士,研究方向:有机化学类课程教学与医药中间体的合成开发。
10.3969/j.issn.2095-4565.2016.04.006
O626
A
2095-4565(2016)04-0021-05
