RDX-Aluminum Interaction-A DFT Study
2016-09-19Lemirker
Lemi Türker
(Department of Chemistry,Middle East Technical University,Ankara Turkey 06231)
RDX-Aluminum Interaction-A DFT Study
Lemi Türker
(Department of Chemistry,Middle East Technical University,Ankara Turkey 06231)
Within the constraints of density functional theory (UB3LYP/6-311++G(d,p)), RDX/Al and RDX/2Al composites are investigated, considering various multiplicity states (singlet and triplet states). Depending on the localization of Al atom(s) in space and multiplicity of the composite systems, the structure of RDX undergoes various degrees of perturbations. It has been shown that the presence of Al atoms affects the bond lengths, electron population as well as the HOMO and LUMO energies and the inter frontier molecular orbital energy gap of RDX. All these perturbations are thought to affect ballistic properties of the explosive molecule RDX.
RDX; aluminum; explosives; DFT calculations; cyclotrimethylene trinitramine; 1,3,5-trinitrohexahydro-sym-triazine.
Introduction
Energetic materials have been getting used extensively for civil as well as military applications. Variety of energetic materials have been synthesized through the decades and used for many purposes. Higher performance is always the prime importance however due to many catastrophic accidents such explosives have become less popular while insensitive explosives appear on the stage in recent years.
RDX (1,3,5-trinitroperhydro-1,3,5-triazine), an important modern molecular explosive, has attracted the attention of scientists for many years since it has been synthesized by Henning in 1899 for medicinal use and used as an explosive in 1920 by Hertz[1].

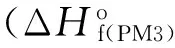
Quite often metals are included in the recipe of ammunitions. One of them is aluminum and it is frequently employed in the formulations of many composite explosives or propellants in order to get better performance of explosives and rocket propellants. Aluminized explosives can be considered as non-ideal explosives because their properties significantly differ than those ones predicted by equilibrium one-dimensional steady state calculations. Reaction temperature and the reaction time can be raised by adding aluminum to explosives, thus the performance of explosives is increased. Some investigators have researched on the role of aluminum powder in the detonation process in explosives[7-10].
Aluminized explosives have enhanced air blast, increased reaction temperature and in underwater systems increased bubble energies. It can be generally assumed that combustion of aluminum particles in the explosion process occurs behind the reaction front during the expansion of gaseous detonation products so that aluminum particles do not participate in the reaction zone, but act as inert ingredients[11-12]. Thermodynamic studies on detonation parameters have been performed by assuming a certain degree of aluminum oxidation[13]. However, it is not clear what degree of aluminum oxidation occurs in the C-J point for mixture of explosives with aluminum.
Aluminized RDX has attracted attention of scientists both experimentally and theoretically[4-6,14-25]. In one of the theoretical works, Carlos et al.[18-19]studied the theoretical infrared and terahertz spectra of an RDX/Al complex by using density functional theory (DFT) approach. In their model RDX is deposited over an Al surface ( planar cluster of Al16). The results indicated that the RDX molecule changed its conformation because of the interaction with Al model surface attaining AAA conformation with the three NO2groups.
In the present study, within the constraints of DFT, interaction of Al and RDX have been studied at the molecular level considering multiplicity of the composite systems.
1 Method of calculation
All the theoretical methods were applied using unrestricted level of theory[26]. The initial geometry optimizations of the RDX/Al composite structures leading to energy minima were achieved by using MM2 method followed by semi-empirical PM3 self-consistent fields molecular orbital (SCF-MO) method[26-27]. Then, further geometry optimizations were achieved using STO and UHF levels of theory (6-31G(d,p)) and then within the framework of Density Functional Theory (DFT, UB3LYP)[27-29]sequentially at the levels of 6-31G(d,p)) and 6-311++G(d,p). The exchange term of B3LYP consists of hybrid Hartree-Fock and local spin density (LSD) exchange functions with Becke′s gradient correlation to LSD exchange[30]. The correlation term of B3LYP consists of the Vosko, Wilk, Nusair (VWN3) local correlation functional[31]and Lee, Yang, Parr (LYP) correlation correction functional[32].
Total electronic energy calculations of all the considered structures have been done at UB3LYP/6-311++G(d,p) theoretical level. The normal mode analysis for each compound yielded no imaginary frequencies, which indicates that each compound has at least a local minimum on the potential energy surface. The total electronic energies were corrected for zero point vibrational energies (ZPVE). All these computations were performed by using Spartan 06 package program at standard conditions of 298.15 K and 1.00×105Pa[33].
2 Results and discussion
The stable conformation of RDX at room temperature is chair AAE (see Fig.1). In the gas phase, electron diffraction experiments reveal that RDX has a chair AAA (C3v) symmetry[34]. In the solid phase RDX has two polymorphs. Theα- RDX is stable at room temperature (orthorhombic Pbca space group) and has a chair AAE structure[35]. Theβ-RDX is extremely unstable at room temperature[36].
In the present study, RDX-aluminum interaction in vacuum conditions has been studied within the limitations of the DFT level of B3LYP/6-311++G(d,p).

Fig.1 The most stable conformer (AAE) of RDX at the room temperature (B3LYP/6-311++G(d,p),hydrogens are not shown)
2.1Structures
Presently, the RDX and aluminum composite systems having mole ratios of RDX/Al and RDX/2Al have been considered. The RDX/Al composite constitutes a doublet system (1) due to the electronic configuration of aluminum atom (1s22s22p63s23p1)[37].Whereas for RDX/2Al composite system, theoretically it is possible to construct singlet (2) and triplet structures (3 and 4). Fig.2 shows the optimized structures and dipole moment vectors of them. The present optimization studies enabled to capture two triplet structures (See Fig.2). Some bond lengths and atom-atom distances of these four systems are shown in Fig.3.
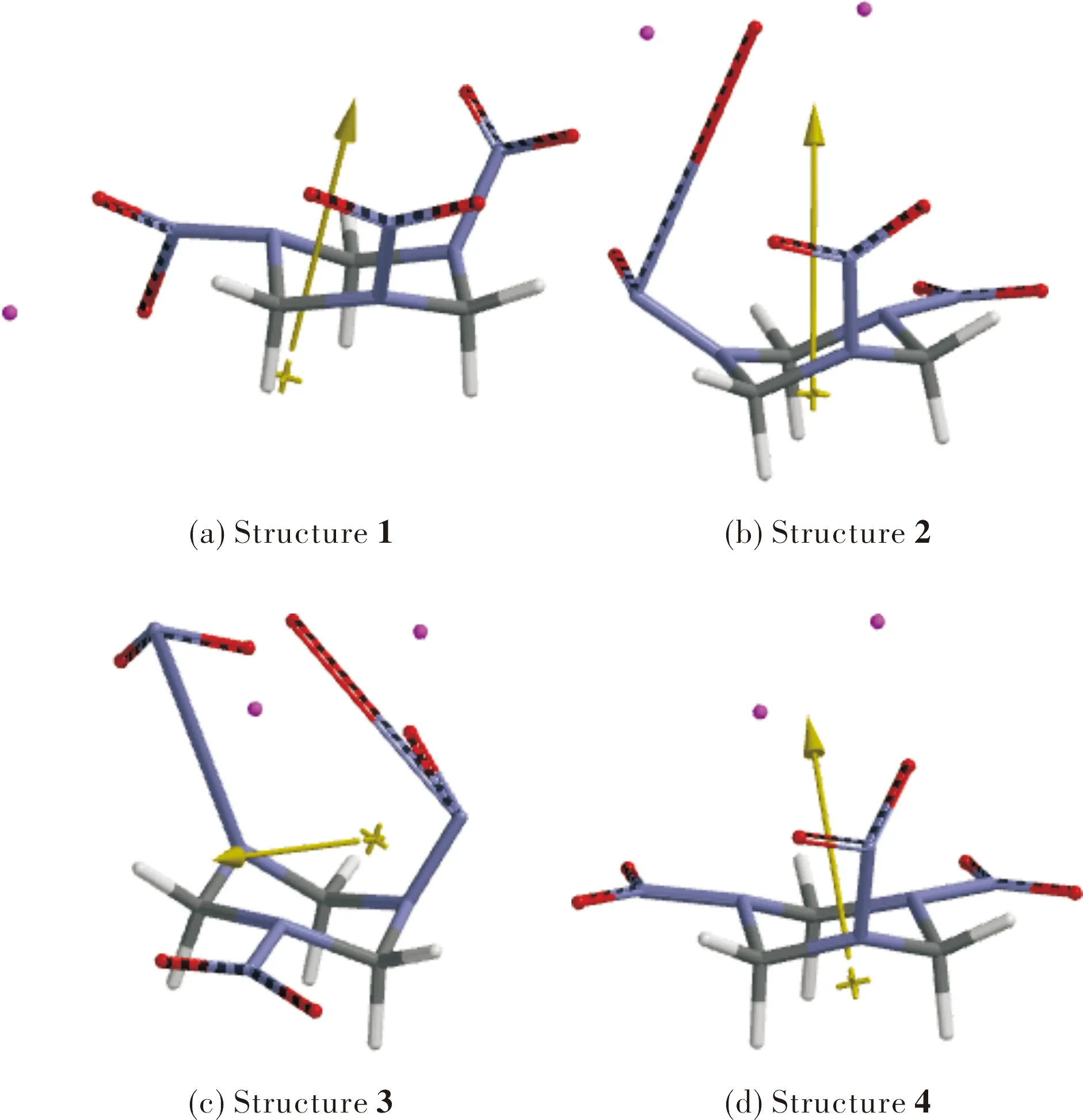
Fig.2 Optimized structures of the composites considered.

Fig.3 The bond lengths/ distances (Å) in various RDX/Al composites (hydrogens are not shown)
The calculations indicate that RDX/Al composite seems to be stable whereas RDX/2Al composites with the exception of triplet structure 4 undergo bond cleavage(s).
In singlet structure 2, one of the N-O bonds of a nitro group is broken (the nitrogen-oxygen distance is 4.05 Å). In Structure 3, one of the NO2group possesses elongated (broken) N-O linkage (3.88 Å). Also in Structure 3 N-NO2bond undergoes bond cleavage (the distance is 3.59 Å) (see Fig.2 and 3).
Fig.4 shows the electrostatic charge distribution in the composites presently considered. In composites 1,3 and 4, Al atom acquires certain degree of positive charge development. In composite 3 the aluminum atoms gets 1.4 and 0.645 unit of charge, indicating electron transfer from Al atoms to RDX system which is partially reduced. In composite 2 Al atoms are almost neutral but one has a minute negative charge development. Whereas the nearby oxygen atom of nitro group gets positive charge, implying slight tendency to form a complex between aluminum and oxygen atoms.
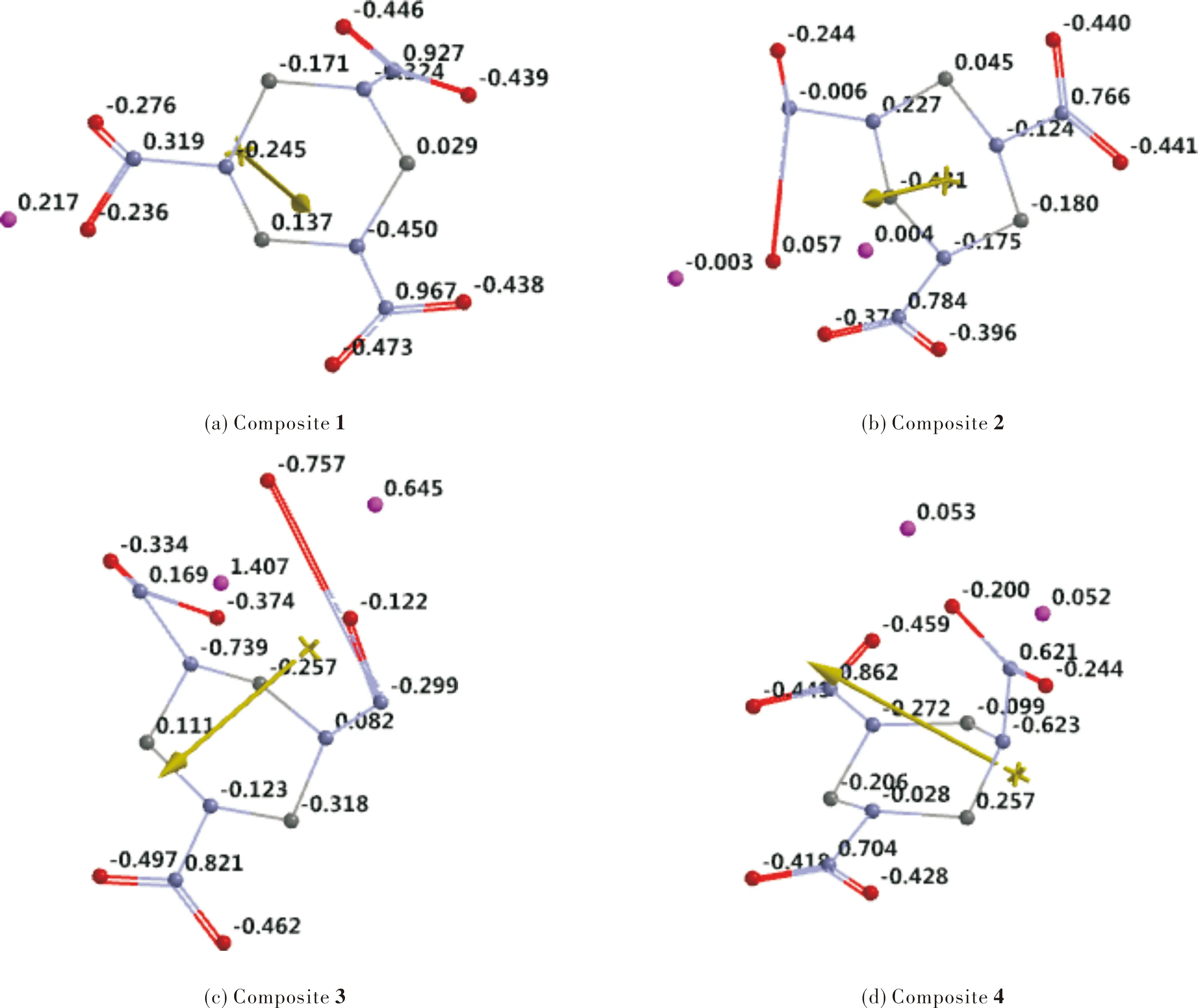
Fig.4 Electrostatic charges in composites 1-4 (hydrogens are depleted)
RDX/Al doublet (1) and RDX/2Al triplet (4) structures remain intact in the optimization process. The nitro groups in structure 1 and 4 , have chair AAE and chair EEA conformations, respectively. Note thatα-RDX also has AAE conformation. The MP2 and DFT/B3LYP calculations by Rice and Chabalowski indicated that AAE conformer was the most stable and EEE was the least stable one[38]. Al-Al distances in RDX/2Al composites (2-4) are 3.46, 2.71 and 2.92 Å, respectively. Structures 2 (singlet) and 3 (triplet) seem to be initially AAA and AAE conformers of RDX, respectively. The optimization results then indicate that those conformers cannot remain intact in the presence of 2Al atoms having singlet and triplet multiplicity, respectively. Table 1 shows some properties of the composites considered. The dipole moments of 1 and 2 are comparable. Structure 3 has the highest and 4 has the lowest dipolemoment, respectively. Note that both 3 and 4 are triplets. The variations are due to charge distributions and bond lengths /distances in the composites.

Table 1 Some properties of the composites considered
Comparison of the bond length data (same level of calcu-lations) depicted in Figures 1 and 3 for AAE conformation of RDX indicates that the presence of Al atom causes contraction of all the nitramine bonds whereas N-O bonds of NO2group nearby the Al atom are elongated somewhat.
Structures 1 and 2-4 have ca.10% and 20% Al, respectively. For those percentages the reported experimental detonation velocity values for aluminized RDX composites are 8.03 and 7.77km/s, respectively[11]. The present calculations indicate that the conformers of the RDX and the multiplicity of the system are also important in various properties.
2.2Energies
The total energies of the composites presently considered are shown in Table 2. The stability order of RDX/2Al composites follow 2>3>4 sequence. The singlet RDX/2Al (2) is the most stable composite among the composites having the same composition. However, 2 and 3 have ruptured RDX systems.

Table 2 Various energies of the composite systems considered
2.3Molecular orbitals
Table 3 shows the HOMO , LUMO energies and inter frontier energy gaps of the composites, Figure 5 displays some of the molecular orbital energy levels of the systems considered. Note that systems with odd number of electrons inevitable possessα-andβ- types of molecular orbitals in unrestricted molecular orbital treatments (composites 1,3 and 4). Fig.6 depicts the HOMO and LUMO of the composites.

Table 3 The HOMO, LUMO energies and inter frontier energy gaps of the composites
When the molecular orbital energies of two intact composites 1 (doublet) and 4 (triplet) are compared, it is observed that the presence of the second aluminum atom appreciably raises theα-HOMO energy of 4 while the energies ofα- andβ- LUMOs remain almost the same (Fig.5). The HOMO energy order is 4>2>3>1, namely 4 has the highest and 1 has the lowest lying HOMO, respectively. The order indicates that the second Al atom raises up the HOMO energy levels in 2-4 as compared to composite 1 which possesses a single Al atom. Then, 4 is the most and 1 is the least potent composite to undergo oxidation. This situation should arise partly from partial electron transfer and partly from orbital-orbital interactions. Whereas the LUMO energy order is 4>2>1>3. Consequently, 3 should be more susceptible to reductions[39- 40]. The HOMO-LUMO energy gap (interfrontier energy gap,ELUMO-EHOMO) follows the order of 4<2<3<1.

Fig.5 Some of the molecular orbital energies of the composites considered

Fig.6 The HOMO and LUMO of the composites considered
It is known that as theELUMO-EHOMOgap decreases the impact sensitivity increases[41]. Hence, composite 4 is expected to be the most and composite 1 be the least susceptible one to impact. Thus, it is obvious that not only the content of Al but also the spin states affect the properties of the composites.
2.4Spin densities
Fig.7 shows the spin densities on the composite struc-tures having open shell(s). It is worth mentioning that in 3, one of the Al atom has no spin density indicating that it has become a closed shell system by transferring its odd electron (see Fig.4 for electrostatic charges).

Fig.7 Spin densities on the doublet and triplet composites considered
2.5Electrostatic potential maps
Fig.8 shows the electrostatic potential maps of the composites presently considered. The comparison of the maps standing for the composites 1 and 4 (in which RDX remains intact ) reveals that in the former one negative region located on the axial nitro groups, whereas in 4 nearby the equivatorial nitro groups (4 possesses EEA conformation). In 3 which has a ruptured RDX structure, the negative region coincides with the nitro groups.
Fig.8 Electrostatic potential maps of the composites considered
3 Conclusions
(1)The present results indicate that the presence of aluminum atom(s) nearby a RDX molecule causes various degrees of structural, electronic and orbital perturbations which may affect the chemical as well as the ballistic properties of RDX molecule (beside the thermal effect of aluminum).
(2)Depending on the multiplicity of the composite system considered, the compositions having the same RDX/Al ratio exhibit different behavior.
(3)In the light of present results the conjecture is that in solid state various hot points having different sensitivity potential may exist due to the effect of aluminum.
References:
[1]Akhavan J. The Chemistry of Explosives[M]. London: The Royal Society of Chemistry, 2004.
[2]Zhu W, Xiao J, Zhu W,et al. Molecular dynamics simulations of RDX and RDX-based plastic-bonded explosives[J]. Journal of Hazardous Materials, 2009,164:1082-1088.
[3]Keshavarz M H. Simple relationship for predicting impact sensitivity of nitroaromatics, nitramines, and nitroaliphatics[J]. Propellants, Explosives, Pyrotechnics, 2010, 35:175-181.
[4]De Paz J L G, Ciller J. On the use of AM1 and PM3 methods on energetic compounds[J]. Propellants, Explosives, Pyrotechnics, 1993,18:33-40.
[5]Osmont A, Catoire L, Gökalpi,et al. Ab initio quantum chemical predictions of enthalpies of formation, heat capacities, and entropies of gas-phase energetic compounds[J]. Combustion and Flame, 2007, 151:262-273.
[6]Rice B M, Pai S V, Hare J. Predicting heats of formation of energetic materials using quantum mechanical calculations[J]. Combustion and Flame, 1999,118:445-458.
[7]Keshavarz M H. New method for predicting detonation velocities of aluminized explosives[J]. Combust Flame, 2005,142:303-307.
[8]Trzcin’ski WA, Paszula J, Grys S. Detonation parameters and blast wave characteristics of nitrometan mixed with particles of an aluminum-magnesium alloy[C]∥NTREM (11th). Pardubice:[s.n.],2008:299-307.
[9]Florczak B, Salacin’ski T. Influence of nitro-compounds on aluminized composite propellants[C]∥ NTREM (11th) .Pardubice:[s.n.],2008:531-534,.
[10] Makhov M N. Heat of decomposition of aluminized explosives[J]. Khimicheskaya Fizika, 2000, 19(9) :83-87.
[11] Keshavarz M H. A simple theoretical prediction of detonation velocities of non-ideal explosives only from elemental composition[C]∥New Research on Hazardous Materials (PB. Warey Ed.) NY:Nova Science Pub, 2007:293-310.
[12] Keshavarz M H. Prediction of detonation performance of CHNO and CHNOAl explosives through molecular structure[J]. Journal of Hazardous Materials, 2009,166:1296-1301.
[13] Hobbs M L, Baer M R.Modeling ignition chemistry[C]∥10th Symposium (Int) on detonation. Boston:MA,1993:409-418.
[14] Pei H B, Jiao Q J, Qin J F. Reaction process of aluminized RDX-based explosives based on cylinder test[J]. Explosion and Shock Waves, 2014, 34(5):636-640.
[15] Lewis W K, Rumchik C G, Broughton P B, et al. Time-resolved spectroscopic studies of aluminized explosives: chemical dynamics and apparent temperatures[J]. Journal of Applied Physics, 2012, 111(1):014903/1-014903/6.
[16] Sulimov A A, Ermolaev B S, Sukoyan M K. Blast waves in a cylindrical channel generated by the nonideal detonation of aluminum-teflon-RDX high-density formulations[J]. Russian Journal of Physical Chemistry B, 2012, 6(3):397-403.
[17] Wang C, Chen S, Zhao S,et al. Influence of Al powder on mechanical sensitivity of RDX[J]. Initiators and Pyrotechnics, 2010, (1):32-34.
[18] Carlos G P, Hoz M, Julibeth M,et al. Theoretical infrared and terahertz spectra of an RDX/aluminum complex (correction)[J]. Journal of Physical Chemistry A, 2010, 114(29):7815-7815.
[19] Carlos G P, Hoz M, Julibeth M,et al. Theoretical infrared and terahertz spectra of an RDX/aluminum complex[J].Journal of Physical Chemistry A, 2010, 114(6):2284-2292.
[20] Trzcinski W A, Cudzilo S, Paszula J. Studies of free field and confined explosions of aluminum enriched RDX compositions[J]. Propellants, Explosives, Pyrotechnics, 2007, 32(6):502-508.
[21] Trzcinski W A, Cudzilo S, Szymanczyk L. Studies of detonation characteristics of aluminum enriched RDX compositions[J]. Propellants, Explosives, Pyrotechnics, 2007, 32(5):392-400.
[22] Song H C, Wan Y, Bai H P. Study of mechanism and model for catalyzed combustion of Al-RDX-CMDB propellant[J].Chinese Journal of Energetic Materials, 2004, 12(Suppl. 2):390-395.
[23] Chen W H, Song S Z, Hu Y T,et al. Studies on the behavior of dust explosion of aluminum and RDX powders[J].Chinese Journal of Energetic Materials, 2003, 11(2):91-93.
[24] Zhang H, Wang K, Zhu Z. Theoretical explosion temperature of aluminum-containing RDX obtained by thermochemistry calculation[J].Acta Armamentarii, 2002, 23(1):136-138.
[25] Hu D, Sun Z. Studies on the influence of the aluminum particle size on the micro-behavior of the high-speed reaction of aluminum containing RDX powder[J].Explosion and Shock Waves,1995,15(2):122-8.
[26] Stewart J J P. Optimization of parameters for semi empirical methods II. Appl[J]. Journal of Computational Chemistry,1989, 10:221-64.
[27] Stewart J J P. Optimization of parameters for semi empirical methods I, Method[J].Journal of Computational Chemistry,1989, 10:209-20.
[28] Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Physics Review,1965,140:1133-1138.
[29] Parr R G, Yang W. Density Functional Theory of Atoms and Molecules[M].London:Oxford University Press,1989.
[30] Becke A D. Density-functional exchange-energy approximation with correct asymptotic behavior[J]. Physics Review A, 1988, 38:3098-100.
[31] Vosko S H, Vilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis[J]. Canadian Journal of Physics, 1980, 58:1200-1211.
[32] Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation energy formula into a functional of the electron density[J]. Physics Review B. 1988, 37:785-89.
[33] SPARTAN 06[CP/EB].Irvine:Wavefunction Inc,2006.
[34] Shiskov I F, Et’fimova T L, Vilkov L V. Geometric structure of molecule of 1,3,5-trinitro-1,3,5-triazacyclohexane in the gas-phase[J]. Journal of Structural Chemistry,1992 ,33:34-38.
[35] Choi C S, Prince E. Crystal structure of cyclotrimethylenetrinitramine[J]. Acta Crystallogr B, 1972, 28:2857-2862.
[36] Karpowicz R J, Brill T B. Comparision of the molecular structure of hexahydro-1,3,5-trinitro-5-triazine in the vapor, solution and solid phases[J]. Journal of Chemical Physics,1984, 88:348-352.
[37] Durant P J, Durant B. Introduction to Advanced Inorganic Chemistry[M]. London:Longman, 1970.
[38] Rice B M, Chabalowski C F. Ab initio and nonlocal density functional study of 1,3,5-trinitro-s-triazine (RDX) conformers[J]. Journal of Physical Chemistry A,1997, 101:8720-8726.
[39] Fleming I. Frontier Orbitals and Organic Chemical Reactions[M]. London:Wiley, 1976.
[40] Streitwieser A Jr. Molecular Orbital Theory for Organic Chemists[M]. New York:Wiley, 1961.
[41] Ovens E J. Relationship between impact induced reactivity of trinitroaromatic molecules and their molecular structure[J]. Journal of Molecular Structure (Theochem) 1984, 121:213-220.
10.14077/j.issn.1007-7812.2016.04.002
date:2016-06-01;Revised date:2016-06-17
TJ55;O641Document Code:AArticle ID:1007-7812(2016)04-0012-07
Biography:Lemi Türker(1950-),male,Prof.,Dr.,research field:organic chemistry.E-mail:lturker@metu.edu.tr
