β沸石结构及其催化性能的调控
2016-09-18王闻年袁德林李浩任申勇郭巧霞申宝剑
王闻年,袁德林,李浩,任申勇,郭巧霞,申宝剑
(1中国石油大学(北京)重质油国家重点实验室,CNPC催化重点实验室,北京 102249;2中国石油大学(北京)理学院,北京 102249)
β沸石结构及其催化性能的调控
王闻年1,袁德林1,李浩1,任申勇1,郭巧霞2,申宝剑1
(1中国石油大学(北京)重质油国家重点实验室,CNPC催化重点实验室,北京 102249;2中国石油大学(北京)理学院,北京 102249)
β沸石作为一种重要的酸催化剂,结构和酸性对其催化性质有重要影响。通过两种不同的联合处理方式(酸-焙烧处理、酸-水热处理)对其结构和性质进行了调变,并对改性所得的β沸石进行了低温氮气物理吸附、X射线电子能谱(XPS)和红外(IR)的表征分析,最后以正辛烷为原料在500℃下进行了催化裂化反应性能评价。研究发现,可通过不同的处理方法调变最终改性沸石的比表面积和两种Lew is酸性(L1445和L1455)。经焙烧和酸处理后所得样品较经水热和酸联合处理所得样品具有更好的正辛烷裂化活性。前者较高的BET比表面积和Lew is酸是其催化裂化活性好的原因。通过这两种不同的改性方式,β沸石的B酸和两种L酸酸量及其织构性质得到调变,从而使最终沸石的催化性能产生差异。
β沸石;酸处理;焙烧;水热;催化;酸性
引 言
β沸石是一种重要的高硅三维十二元环孔道沸石,其孔道体系由两个相互垂直的直通孔道和一个曲折孔道组成,孔径分别为 0.57 nm×0.75 nm和0.56 nm×0.65 nm[1]。由于其特殊的孔道结构和酸性质,β沸石被用于许多酸催化反应中,例如烷烃裂化[2-3]、有机物合成[4-6]以及化学品转化等[7-9]。
β沸石不同的后处理方式对其铝物种的结构会有不同影响,最终会影响其催化性能[2,10]。对于氢型β沸石而言,非骨架铝物种和骨架缺陷均可作为Lew is酸中心[11]。在高温下,β沸石中铝羟基会脱水而形成Lew is酸中心[12],部分骨架铝会转变为八面体形式存于沸石骨架中;而某些不稳定骨架位的铝则会因高温作用而从骨架上脱落形成非骨架铝,某些较稳定的骨架位的铝则以扭曲四面体的形式存在于骨架中[13]。研究发现,这些扭曲的四面体铝具有Lew is酸性,通常在吸附水分子后以八面体的形式存在于骨架中,但对沸石进行脱水以后,这些铝的配位状态会恢复为四面体形式,并成为Brønsted酸中心[14]。
沸石中非骨架铝的形式多种多样,而对沸石进行水热处理往往会产生大量的非骨架铝,并对沸石的酸性和孔道产生影响。研究发现,水热处理同时降低了沸石的B酸和L酸酸量,且微孔孔道会因非骨架铝的缩聚而被堵塞,部分酸性中心被非骨架铝覆盖[11,15]。而对沸石进行酸处理,往往会形成部分弱L酸中心,这些L酸多为非骨架铝物种,且其位置大多在B酸中心附近[16-17]。对于β沸石而言,其耐酸性较强,只有高浓度酸才能致其脱铝,而低浓度酸只能起到清理非骨架铝的作用,在一定程度上改变了沸石的B酸酸性。而沸石中铝的状态和位置往往关系着沸石的酸性,不同的改性方法又会影响沸石中铝的状态和分布。
因此,本文的目的在于通过两种不同的联合处理方式——酸-焙烧、酸-水热,对β沸石进行改性,以获得不同酸性质的β沸石,并对所得的改性样品进行结构和催化性质的对比研究。
1 实验材料和方法
1.1样品的制备
β沸石原粉是由前文报道的方法合成[3]。通过对原粉β沸石在80℃下以0.8 mol·L-1氯化铵溶液进行两次交换获得铵型β沸石(NH4β)。后处理改性实验过程称之为酸-水热处理或酸-焙烧处理,具体方法为:在80℃下用0.5 mol·L-1的HCl溶液对NH4β进行酸处理,过滤、洗涤沸石至滤液 pH为7.0左右,之后在120℃条件下干燥12 h;将酸处理后的β沸石(βA0.50)分别进行100%水蒸气(到达指定温度后通入水蒸气)或焙烧处理,并分别命名为βAS0.50(水热温度)和βAC0.50(焙烧温度)。对于未经酸处理而直接水热或焙烧的样品,命名为βS(水热温度)或βC(焙烧温度)。
1.2样品的分析和表征
样品的粉末 X射线衍射(XRD)谱图由荷兰PANalytial X’Pert Powder型X射线衍射仪测定。采用CuKα辐射(波长为0.1541 nm),管电流40 mA,管电压40 kV,步长0.013 (°)·s-1。物相分析的扫描角度范围为5°~35°。测量时,取0.4 g左右的待测样品,用研钵研磨至粘壁之后,在120℃下干燥2 h。将干燥好的样品轻压成表面平整的片进行测量。低温氮气物理吸附由美国 M icromeritics TriSTAR 3020型物理吸附仪完成。待测样品在350℃、真空(10~20 Pa)条件下脱水脱气处理8 h,而对于含有模板剂的样品,则在180℃、真空(10~20 Pa)条件下脱水脱气处理12 h。之后在液氮温度(77 K)下吸附测量。由 BET公式计算得到样品的比表面积,BJH法得到孔径分布,t-plot法获得微孔比表面积和微孔体积。X射线电子能谱(XPS)由Thermo Fisher K-Alpha型仪器测定。样品干燥后在4.9 MPa的压力下压制成片,90℃下真空干燥 24 h后,在5×10-7Pa下进行分析测量。Brønsted酸及两种Lew is酸通过吡啶红外(Py-IR)进行定量[18],待测样品充分研磨之后120℃下干燥2 h,取约0.020 g的样在5 MPa的压力下制成直径为15 mm的自支撑圆片。将压好的圆片干燥称重后置于装有CaCl2窗口的红外真空池中,在400℃、10-4Pa真空条件下脱水脱气处理2 h。将预处理的样品与吡啶蒸气进行吸附平衡20 m in后,在指定温度、10-4Pa真空下脱附1 h。之后在室温下收集相应的红外吸收光谱。采集范围为1400~1700 cm-1。其中在1445和1455 cm-1的吸收峰通过Gauss函数进行拟合[19],计算酸量时采用了相同的参数。样品的催化活性以正辛烷为模型化合物进行评价。将干燥并研磨好的β沸石,在10 MPa压力下压制成片。将压好的片捣碎后过筛,筛分出250~420 μm 样品,在120℃下干燥5 h待测。评价反应温度为500℃,正辛烷在40℃预热并通过氮气流将其饱和蒸气引入催化剂床层;催化剂装填量为50 mg;最终裂化产物通过在线气相色谱仪进行分析。
2 实验结果与讨论
2.1不同后改性方法对β沸石宏观性质的影响
2.1.1织构性质酸-焙烧处理、酸-水热处理改性后所得沸石的XRD谱图如图1所示,改性前后的样品的谱图均为典型的 β沸石衍射峰。位于2θ=22.30°附近的衍射峰强度在经过改性后明显下降,说明发生了骨架结构破坏。从表1中样品的结晶度数据可以看出,酸处理对样品的结晶度影响远小于水热和焙烧对样品结晶度的影响。最终样品的结晶度较原粉下降了10%左右。
酸-焙烧处理、酸-水热处理改性后所得沸石的低温氮气物理吸附脱附数据见表1。经过酸处理后,样品βA0.50的比表面积和微孔体积增加明显,但没有达到最终改性样品的水平,说明酸处理仅除去了堵塞在孔道中的部分模板剂。酸溶液起到了萃取孔道中弱作用模板剂的作用[20]。最终改性后沸石的比表面积和孔体积迅速增加是因为沸石中模板剂的完全脱除。酸-焙烧处理样品的微孔表面积和孔体积显著高于酸-水热处理样品的微孔表面积和孔体积,说明水热过程造成了更为严重的沸石骨架破坏。另外,水热形成的非骨架铝物种堵塞了沸石孔道也是造成其样品比表面积较低的原因。各改性样品的Vtotal-Vmicro值均在0.300 cm3·g-1左右,说明不同改性方式对沸石的孔体积的影响不是很大。
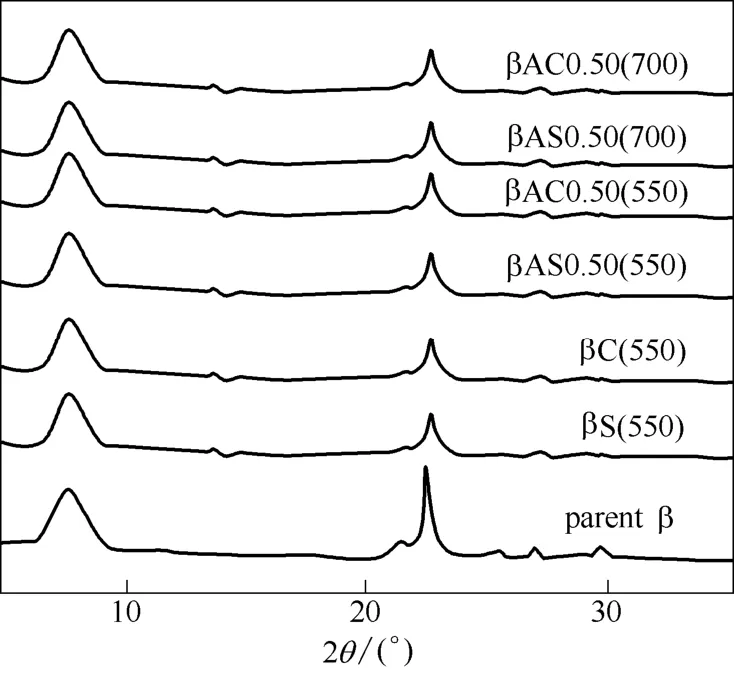
图1 β沸石原粉及经不同处理方法后所得样品的XRD谱图Fig.1 XRD patterns of parent β and β-zeolite obtained after different treatments
图2为各样品的氮气吸附等温线和孔分布曲线。等温线[图2(a)]在相对压力0.80~0.95之间变得陡峭,说明样品中存在介孔结构[21]。对比β沸石原粉和处理后沸石样品的孔分布曲线[图 2(b)],可以发现在9 nm附近均有最可几孔径分布,而焙烧或水热过程使其强度增加,说明该过程中这部分孔的量在增加。由此可以推断,此处的孔因归属于晶粒间的堆积孔,且有部分模板剂吸附在沸石外表面。经过处理以后,堆积孔内的模板剂被脱出,孔体积增加1倍以上。另外,经过改性的样品在87 nm附近出现了大孔分布。
2.1.2后处理前后β沸石的形貌通过图3(a)、(b)中β沸石原粉的扫描电镜(SEM)图片可以看出,β沸石颗粒是由50 nm左右的晶粒组成的1~3 μm的团聚体,而在孔分布曲线上出现的9 nm孔分布应来自于这些纳米晶粒之间的粒间孔,而87 nm处的大孔应该来自更大颗粒的堆积孔。经过处理以后[图 3(c)~(h)],样品中部分大颗粒变小,形成了较多的小颗粒,使颗粒整体表面形貌变得更为粗糙。
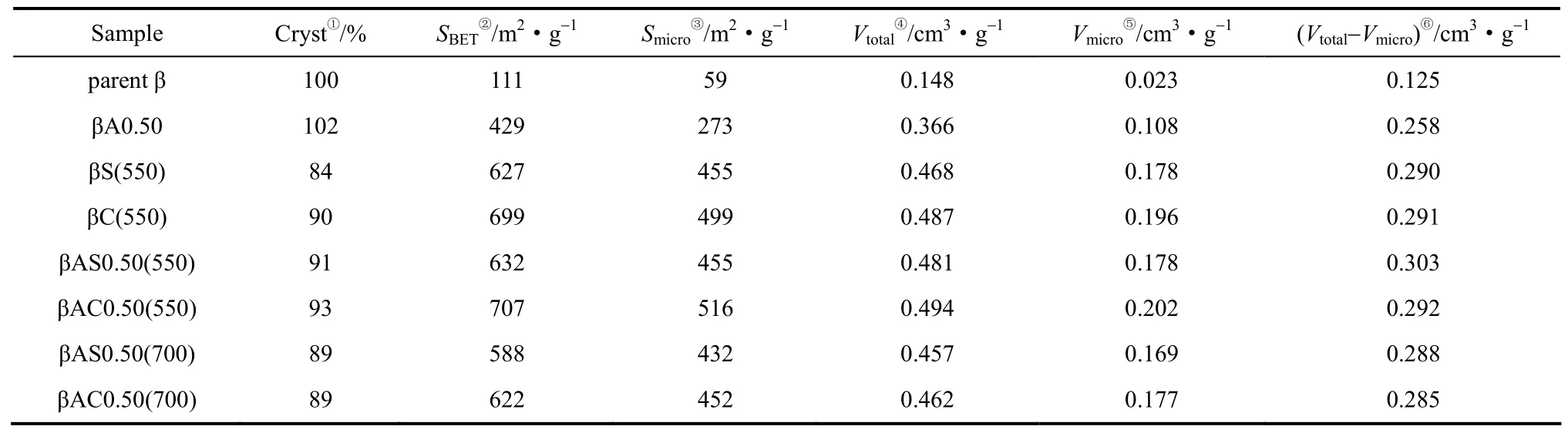
表1 β沸石的织构性质Table 1 Textural properties of β-zeolites
2.1.3样品的元素组成表2为各β沸石样品通过XPS能谱获得的表相元素组成(硅铝摩尔比)、Si2p与 Al2p的电子结合能和半峰宽数据以及红外光谱中骨架四面体不对称振动波数(νTOT)。
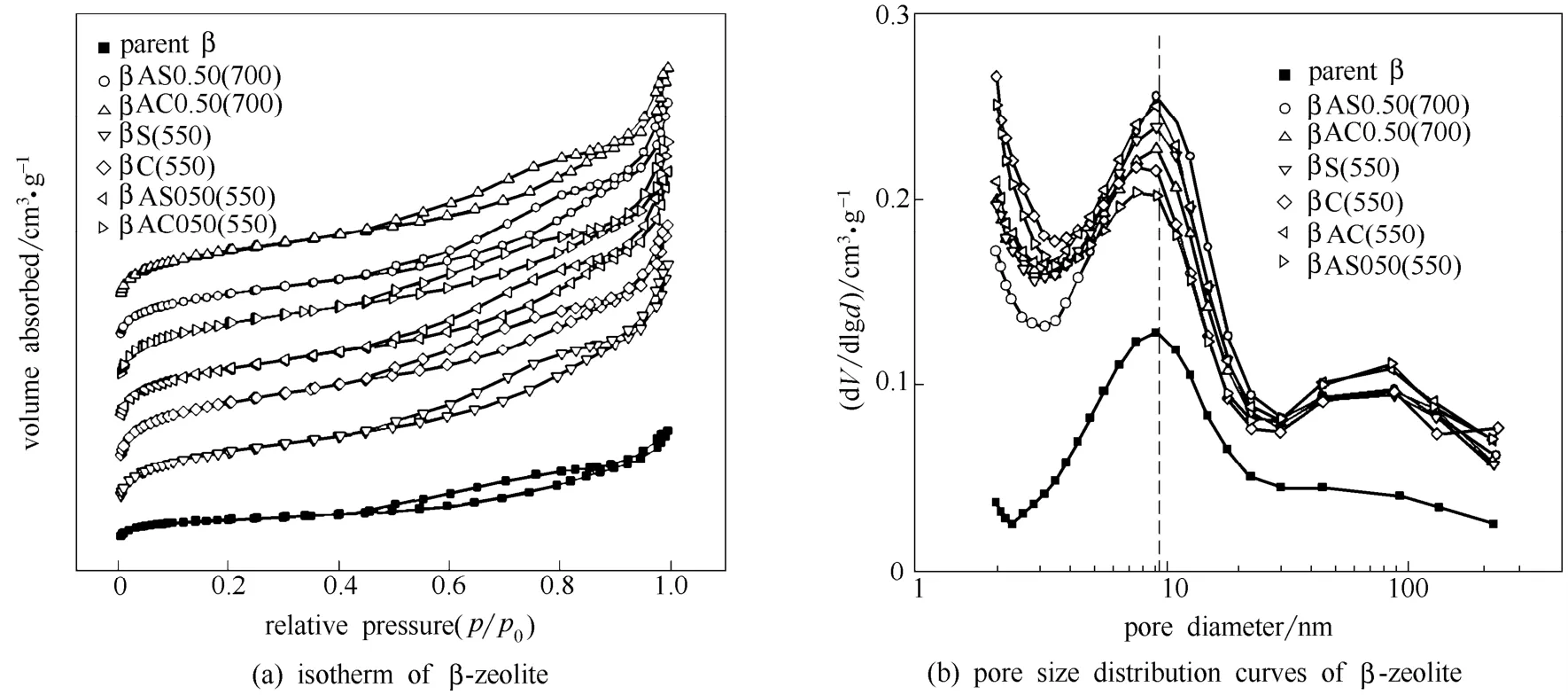
图2 β沸石原粉、βS(550)、βC(550)、βAS0.50(550)、βAC0.50(550)、βAS0.50(700)、βAC0.50(700)的低温氮气吸附等温线和孔分布曲线Fig.2 Isotherm of N2adsorption and desorption and pore size distriobution curves of parent β, βS(550), βC(550), βAS0.50(550),βAC0.50(550), βAS0.50(700), βAC0.50(700)

可以看出,除了样品βS(550)和βC(550),其余样品的表相SiO2/Al2O3均有增加,说明表相铝原子在改性过程中被脱除。因为酸处理的是含有模板剂的沸石,孔道中的模板剂阻止了氢离子的进入,酸处理只能将表面的部分铝原子脱除。样品 βS(550)的表面SiO2/A l2O3最低,是因为水热过程有非骨架铝迁移到沸石表面[22]。Si2p电子结合能在处理前后没有很大变化,A l2p电子结合能则在处理后略有增加。对于β沸石原粉和样品βA0.50,其FWHM(Al2p)/ FWHM(Si2p)值低于1,说明原粉中铝均以四面体形式存在于骨架中[23];经改性后该比值增加,且骨架不对称振动波数(νTOT)增加,说明样品发生了显著的骨架脱铝[11]。样品βAS0.50(700)和βAC0.50(700)具有相同的骨架不对称振动波数说明两者具有相当的脱铝程度,而βAS0.50(550)较βAC0.50(550)该值较高,说明水热在较低温度下就能达到较高程度的脱铝。温度在700℃时,两者之间的差异消失。

图3 β沸石原粉和β沸石的扫描电镜(SEM)图片Fig.3 Scanning electron m icroscope (SEM) images of parent β and β-zeolites

表2 β沸石的XPS SiO2/A l2O3、Si2p与A l2p电子结合能(BE)、半峰宽(FWHM)值及骨架不对称振动波数(νTOT)Table 2 XPS SiO2/A l2O3, binding energies (BE), full w idths at half-maximum (FWHM) of Si2p and Al2p peaks,and framework structure asymmetric stretch vibration wavenum bers (νTOT) of β-zeolites
2.2不同后处理方法对β沸石酸性质的影响
图4为所得样品的Py-IR谱图,在1445 cm-1(L1445)附近出现的吸收峰归属为吸附在Lew is酸中心并与邻近的羟基有键合的吡啶分子振动吸收峰[17,24],而在1455 cm-1(L1455)附近出现的吸收峰归属为吸附在Lew is酸中心的吡啶产生的振动吸收峰。位于1540 cm-1附近的吸收峰为吡啶吸附在B酸中心所产生的吸收峰。表3为通过Py-IR实验计算所得的不同处理方法所得样品的 B酸和 L酸浓度。可以看出,水热处理样品的B酸中心少于焙烧处理样品的B酸中心,说明水热导致了更多骨架铝被脱除,这与表2中νTOT所得的结果一致。
当有酸处理时,水热处理样品中有更多的L1445酸中心,这部分L1445酸中心与羟基邻近,但是接近性较差[25]。这部分酸中心仅仅是一个三配位骨架铝原子[24],亦即骨架缺陷位。可见,酸-水热处理形成了更多的骨架缺陷位,这与水热处理样品具有较低的比表面积相对应(表1)。而焙烧样品则具有更多的L1455酸中心,这部分酸中心多为非骨架阳离子铝物种形成的L酸酸中心。从350 ℃的吡啶脱附数据可以看出,大部分L1445为弱酸中心。而大部分L1455为强酸中心。
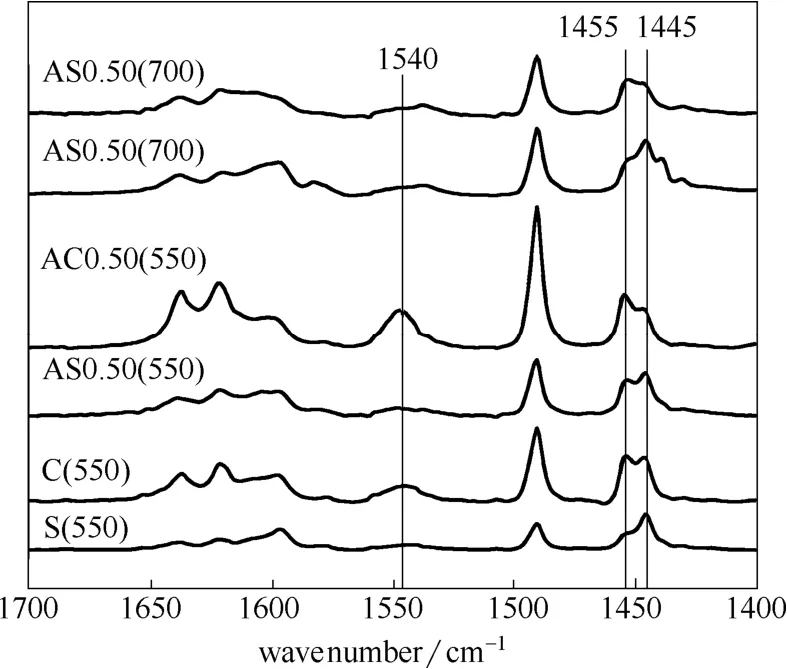
图4 样品的吡啶红外谱图Fig.4 Pyridine adsorbed IR spectra of β-zeolites
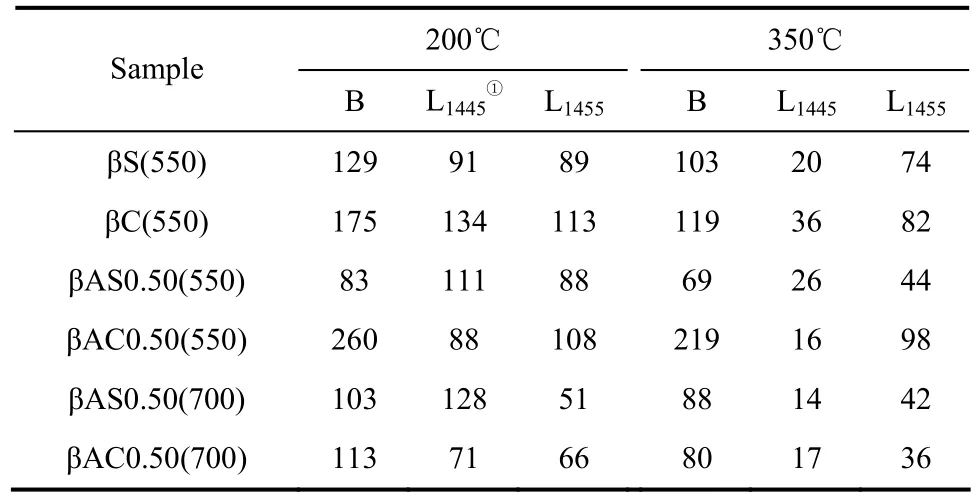
表3 不同处理条件下所得β沸石的吡啶红外数据Table 3 Concentration of Brønsted acid sites and Lew is acid sites of β-zeolites/μmol·g-1
当没有酸处理时,焙烧形成了更多的L1445酸中心,这可能是因为没有酸处理除去沸石表面和孔道中的部分模板剂,导致焙烧过程产生的原位水蒸气造成了大量骨架缺陷。对于水热过程,可能是因为在升温过程中原位水蒸气形成了缺陷,而在后续水热过程中水蒸气在缺陷位进一步发生脱铝。从表 2可以看出,其表相硅铝比较低,说明表面富铝,更多铝原子已完全从骨架上脱除并迁移至外表面,因此骨架缺陷较少。
不论有无酸处理,焙烧处理产生了更多的L1455酸中心。但水热导致了更多的骨架脱铝,较低的L1455酸中心浓度应与水热过程中有非骨架物种聚合有关[17]。
对于 550℃下处理的样品,焙烧处理所得样品的B酸量远高于水热处理所得样品的B酸酸量;在700℃下,焙烧处理所得样品的 B酸量略高于水热处理所得样品的B酸酸量。不同温度下产生的L1445酸酸量相差不大,而低温处理形成了更多的 L1455酸中心。
2.3不同后处理方法对β沸石催化活性的影响
图5为样品βS(550)、βC(550)、βAS0.50(700)以及βAC0.50(700)在500℃下的正辛烷催化裂化转化率。可以看出,焙烧改性样品的正辛烷转化率显著高于水热改性样品的正辛烷转化率。烷烃的裂化与催化剂的B酸中心数目有关[2],从表3可以看出焙烧样品的B酸中心数目高于水热样品的B酸中心数目。样品βC(550)因B酸中心数较高,随着反应时间的延长,形成大量积炭,正辛烷的转化率逐渐下降。而样品βAC0.50(700)酸量适宜,催化裂化活性没有随着反应时间的延长而降低。
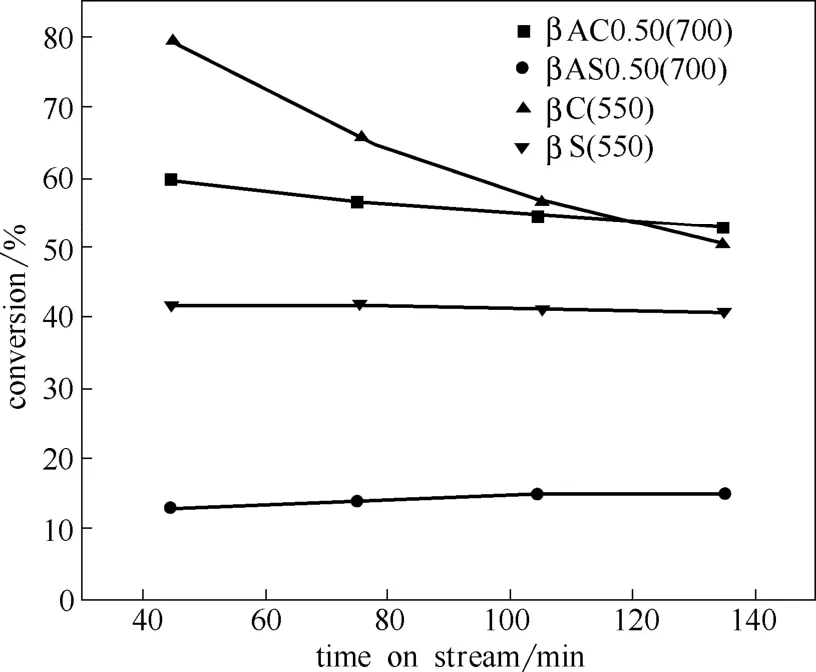
图5 样品在500℃下的正辛烷转化率Fig.5 Conversion of n-octane cracking of samples at 500℃
而样品 βS(550)的 B 酸酸量高于样品βAC0.50(700)的B酸酸量,但其正辛烷转化率却较低。因此,可以推断,酸-焙烧样品较高的转化率不仅来自于质子酸裂化[2],还应归因于在B酸中心附近的L酸对B酸酸性强度的增强作用[26-27]。这部分L酸应为以非骨架阳离子铝形式存在的 L1455酸中心。很有可能是因焙烧形成的这些非骨架铝物种没有迁移,更接近B酸中心,从而增强了B酸酸性,提高了样品βAC0.50(700)的正辛烷裂化能力。另外,酸-焙烧样品较高的比表面积也是其转化率较高的原因之一。
3 结 论
通过酸-水热处理和酸-焙烧处理的方法可以调节β沸石的比表面积和L酸中心的分布。酸处理除去了堵塞在孔道中的部分模板剂并脱除了表面的部分铝物种。后续的水热处理过程较焙烧过程相比,形成了更多的沸石骨架缺陷位,且有更多的非骨架铝物种迁移到沸石表面。β沸石的脱铝程度和非骨架铝的形成与两种热处理(焙烧和水热)的温度也有关,较低的水热温度就可以达到较好的骨架脱铝。焙烧形成的非骨架铝物种没有发生迁移,形成的非骨架铝阳离子物种增强了其附近的B酸酸性,因此有较高的烃类催化裂化活性。最终酸-焙烧处理方法得到的样品具有较高的比表面积和较强的B酸-L酸协同作用,其催化性能优于酸-水热处理方法所得样品的催化性能。通过不同的联合处理方法调变沸石的结构和酸性,从而可对其催化性能进行针对性调控。
References
[1]HIGGINS J B, LAPIERRE R B, SCHLENKER J L, et al. The framwork topology of zeolite β [J]. Zeolites, 1988, 8: 446-452.
[2]MARQUES J P, GENER I, LOPES J M, et al. n-Heptane cracking on dealum inated HBEA zeolites [J]. Catal. Today, 2005, 107/108: 726-733.
[3]SHEN B J, WANG P, ZHOU Y, et al. Synthesis of zeolite β from kaolin and its catalytic performance for FCC naphtha aromatization [J]. Energy & Fuels, 2009, 23: 60-64.
[4]HEINICHEN H K, HOLDERRICH W F. Acylation of 2-methoxynaphthalene in the presence of modified zeolite HBEA [J]. J. Catal., 1999, 185: 408-414.
[5]CASAGRANDE M, STORARO L, LENARDA M, et al. Highly selective Friedel-Crafts acylation of 2-methoxynaphthlene catalyzed by H-BEA zeolite [J]. Appl. Catal. A Gen., 2000, 201: 263-270.
[6]ZHU Y Z, CHUAH G, JAENICKE S. Chemo- and regioselective Meerwein-Ponndorf-Verley and Oppenauer reactions catalyzed by A l-free Zr-zeolite β [J]. J. Catal., 2004, 227: 1-10.
[7]LIU N, ZHANG R D, CHEN B H, et al. Comparative study on the direct decomposition of nitrous oxide over M (Fe, Co, Cu)-BEA zeolites [J]. J. Catal., 2012, 294: 99-112.
[8]BATALHA N, PINARD L, BOUCHY C, et al. n-Hexadecane hydroisomerization over Pt-HBEA catalysts. Quantification and effect of the intimacy between metal and protonic sites [J]. J. Catal., 2013,307: 122-131.
[9]OGURA M, ITABASHI K, DEDECEK J, et al. Stabilization of bare divalent Fe(Ⅱ) cations in A l-rich β zeolites for superior NO adsorption [J]. J. Catal., 2014, 315: 1-5.
[10]KUNKELER P J, ZUURDEEG B J, VAN DER WAAL J C, et al. Zeolite β: the relationship between calcination procedure, alum inum configuration, and Lew is acidity [J]. J. Catal., 1998, 180: 234-244.
[11]MARQUES J P, GENER I, AYRAULT P. Semi-quantitative estimation by IR of framework, extraframework and defect Al species of HBEA zeolites [J]. Chem. Commun., 2004: 2290-2291.
[12]CREYGHTON E J, GANESHIE S D, DOWNING R S, et al. Stereoselective Meerwein-Ponndorf-Verley and Oppenauer reactions catalysed by zeolite BEA [J]. J. Mol. Catal. A Chem., 1997, 115: 457-472.
[13]JIA C, MASSIANI P, BARTHOMEUF D. Characterization by infrared and nuclear magnetic resonance spectroscopies of calcined β zeolite [J]. J. Chem. Soc. Faraday Trans., 1993, 89: 3659-3665.
[14]DE MENORVAL L C, BUCKERMANN W, FIGUERAS F. Influence of adsorbed molecules on the configuration of framework alum inum atoms in acidic zeolite-β. A27Al MAS NMR study [J]. J. Phys. Chem., 1996, 100: 465-467.
[15]BATONNEAU-GENER I, YONLI A, SOLANGE H, et al. Influence of steaming and acid-leaching treatments on the hydrophobicity of HBEA zeolite determ ined under static conditions [J]. M icroporous Mesoporous Mater., 2008, 110: 480-487.
[16]M IRODATOS C, BATTHOMEUF D. Superacid sites in zeolites [J]. J. Chem. Soc. Chem. Commun., 1981: 39-40.
[17]MARQUES J P, GENER I, AYRAULT P, et al. Infrared spectroscopic study of the acid properties of dealum inated BEA zeolites [J]. M icroporous Mesoporous Mater., 2003, 60: 251-262.
[18]EMEIS C A. Determ ination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts [J]. J. Catal., 1993, 141: 347-354.
[19]AHN J H, KOLVENBACH R, NEUDECK C, et al. Tailoring mesoscopically structured H-ZSM-5 zeolites for toluene methylation [J]. J. Catal., 2014, 311: 271-280.
[20]JONES C W,TSUJI K, TAKEWAKI T, et al. Tailoring molecular sieve properties during SDA removal via solvent extraction [J]. M icroporous Mesoporous Mater., 2001, 48: 57-64.
[21]XIAO F S, WANG L F, YIN C Y, et al. Catalytic properties of hierarchical mesoporous zeolites templated w ith a m ixture of small organic ammonium salts and mesoscale cationic polymers [J]. Angew. Chem. Int. Ed., 2006, 45: 3090-3093.
[22]JANSSEN A H, KOSTER A J, DE JONG K P. Three-dimensional transm ission electron m icroscopic observations of mesopores in dealum inated zeolite Y [J]. Angew. Chem ie Int. Ed., 2001, 40: 1102-1104.
[23]COLLIGNON F, JACOBS P A, GROBET P, et al. Investigation of the coordination state of aluminum in β zeolites by X-ray photoelectron spectroscopy [J]. J. Phys. Chem. B, 2001, 105: 6812-6816.
[24]VIMONT A, THIBAULT-STARZYK F, LAVALLEY J C. Infrared spectroscopic study of the acidobasic properties of β zeolite [J]. J. Phys. Chem. B, 2000, 104: 286-291.
[25]ONG L H, DÖMÖK M, OLINDO R, et al. Dealum ination of HZSM-5 via steam-treatment [J]. M icroporous Mesoporous Mater.,2012, 164: 9-20.
[26]LI S H, ZHENG A, SU Y C, et al. Brønsted/Lew is acid synergy in dealum inated HY zeolite: a combined solid-state NMR and theoretical calculation study [J]. J. Am. Chem. Soc., 2007, 129: 11161-11171.
[27]ABREVAYA H. Cracking of naphtha range alkanes and naphthenes over β-zeolites [J]. Stud. Surf. Sci. Catal., 2007, 170: 1244-1251.
Regulation of structure and catalytic performance of β-zeolite by post treatments
WANG Wennian1, YUAN Delin1, LI Hao1, REN Shenyong1, GUO Qiaoxia2, SHEN Baojian1
(1State Key Laboratory of Heavy Oil Processing, the Key Laboratory of Catalysis of CNPC, China University of Petroleum,Beijing 102249, China;2College of Science, China University of Petroleum, Beijing 102249, China)
β-Zeolite, an important acidic catalyst, its structure and acidity can significantly influence the catalytic activity. Through two combinational treatments of acid-calcination and acid-steaming, the structure and acidity of β-zeolite were tuned. After treatment, the modified β-zeolites was characterized by N2adsorption and desorption,X-ray photoelectron spectroscopy (XPS), and infrared spectra (IR). The activity for catalytic cracking of n-octane at 500℃ was evaluated. It showed that BET specific area and Lew is acid sites (L1445and L1455) were tuned by different treatments. The catalyst by acid-calcination treatment exhibited better activity for catalytic cracking of n-octane than acid-steaming treatment. The higher BET surface area and the Lew is acid sites account for the higher catalytic cracking activity of the catalyst by acid-calcination treatment. The concentrations of Brønsted acid and two types of Lew is acid as well as textural properties were tuned, which lead to the difference in catalytic activity between the original and final β-zeolites.
β-zeolite; acid treatment; calcination; steam ing; catalysis; acidity
date: 2016-03-24.
Prof. SHEN Baojian, baojian@cup.edu.cn
supported by the National Basic Research Program of China (2012CB215001) and the National Natural Science Foundation of China (U1462202).
TE 624.9
A
0438—1157(2016)08—3429—07
10.11949/j.issn.0438-1157.20160340
2016-03-24收到初稿,2016-04-25收到修改稿。
联系人:申宝剑。第一作者:王闻年(1986—),男,博士研究生。
国家重点基础研究发展计划项目(2012CB215001);国家自然科学基金项目(U1462202)。