氧化石墨烯复合磷酸酯交联弹性体的制备及其用于药物缓释的研究
2016-09-18孙黄辉徐志珍张黎伟张文清
孙黄辉, 徐志珍, 夏 玮, 张 姝, 张黎伟, 张文清
(1.华东理工大学上海市功能性材料化学重点实验室,上海 200237; 2.云南林源香料有限公司,云南玉溪 651100)
氧化石墨烯复合磷酸酯交联弹性体的制备及其用于药物缓释的研究
孙黄辉1,徐志珍1,夏玮1,张姝1,张黎伟2,张文清1
(1.华东理工大学上海市功能性材料化学重点实验室,上海 200237; 2.云南林源香料有限公司,云南玉溪 651100)
选取亲水性较好的聚乙二醇,通过自身的相转移催化性进行环氧化,制备的环氧化产物与聚四氢呋喃磷酸酯和氧化石墨烯进行混合、交联固化,制备了可降解的磷酸酯交联材料。结果表明制备的交联材料具有良好的力学性能和降解性能,氧化石墨烯的添加影响交联材料的力学性能和降解性能。体外的药物释放测试和细胞毒性研究表明制备的聚合物作为药物释放体系具有较大的应用潜力。
氧化石墨烯; 磷酸酯键; 生物材料; 药物载体
氧化石墨烯(GO)由于具有独特的物理化学性质,在生物医学领域引起了人们越来越多的关注[1-3]。其中,GO较高的比表面积和大量的sp2杂化碳原子区域等固有的性质,使其在药物释放方面具有巨大的潜质。Dai等[4]首先利用π-π相互作用将阿霉素负载到聚乙二醇功能化的氧化石墨上,实验表明此载药体系具有良好的生物相容性,能有效地实现药物的搭载和释放。随后,有文献[5]报道了水溶性较差的喜树碱模型药物SN38,通过π-π作用搭载到GO表面,达到了在水溶液中可持续释放的目的。此后报道了大量的关于GO进行药物搭载和释放的研究[6-8],都取得了不错的效果。GO表面具有大量的羟基、羰基、羧基等含氧基团,使其易于与聚合物结合,同时,因其具有特殊的力学性能,因此近年来被作为纳米增强填料来制备复合材料[9]。溶液共混法因其操作简单是目前使用最广泛的制备GO/聚合物复合材料的方法,常用来制备聚氨酯、聚苯乙烯等复合材料,但是其聚合结束后溶剂很难除去的缺点限制了其在生物医学领域的应用[10]。
聚乙二醇(PEG)具有较强的亲水性,较低的生物毒性以及较易修饰的特点[11-12],广泛应用于生物医学领域。由于PEG本身难以形成交联结构[13],通常将其端羟基进行活性官能团的修饰,利用官能团之间的反应,引入交联材料中。但是传统的制备PEG交联材料的方法形成的交联位点多为不具备明显水解性和酶解性的烷基或者氨基甲酸酯等,大大限制了其在生物体内的实际应用[14-15]。为了提高PEG交联材料降解性而引入的酯键、二硫键以及肽键等虽然有效地提高了材料的降解性,但是在合成过程中不可避免地引入了一些有毒的催化剂或引发剂等,因而对细胞具有较强的毒性[16]。磷酸酯是一种具有良好水解性和酶解性的化学键,广泛存在于生物体内。自从20世纪70年代,Penczek等[17]研究了磷酸酯的反应机理以来,越来越多的科研人员对其产生了浓厚兴趣。
本文根据Kubisa等[18]制备磷酸酯键的方法,在无任何添加剂的条件下混合不同分子量的环氧化的聚乙二醇(PEG-E)和磷酸化的聚四氢呋喃二醇(PTMEG-P),通过掺杂不同量的GO在无溶剂的条件下制备了不同性能的交联材料。降解性测试和毒性测试证明了制备的材料是一种可以完全降解,具有较低细胞毒性的交联弹性体。物理包裹的药物释放实验表明制备的材料具有一定的药物缓释功能,特别是添加了GO的交联材料具有更低的细胞毒性和更好的药物缓释功能,说明制备的交联材料在生物医学领域具有一定的应用前景。
1 材料与仪器
膨胀石墨(上海一凡实业有限公司),聚四氢呋喃(数均分子量为1 000,阿拉丁试剂有限公司),聚乙二醇(数均分子量为200、400、600、1 000,国药集团化学试剂有限公司),环氧氯丙烷(分析纯,国药集团化学试剂有限公司),多聚磷酸(分析纯,国药集团化学试剂有限公司),五氧化二磷(分析纯,国药集团化学试剂有限公司)。
万能试验机4302(布鲁克(北京)科技有限公司),DD2400-MR核磁共振仪(美国安捷伦有限公司),METTLER AE240型分析天平(梅特勒-托利多有限公司),Nicolet380傅里叶变换红外光谱仪(赛默飞世尔科技有限公司),CO2细胞培养箱(美国 Thermo),UV-2550紫外可见分光光度计(岛津仪器有限公司)。
2 实验方法
2.1氧化石墨烯(GO)的制备
根据改进的Hummers法[19],通过浓磷酸、浓硫酸和高锰酸钾对膨胀石墨进行氧化,经过去离子水洗涤干燥后完成GO的制备,得到黑色粉末状固体,超声后达到分子水平的分散。
2.2磷酸化聚四氢呋喃(PTMEG1000-P)和环氧化聚乙二醇(PEG-E)的制备
选取分子量为1 000的聚四氢呋喃二醇(PTMEG1000)作为原料,利用多聚磷酸及五氧化二磷作为混合磷酸化试剂制备PTMEG1000的磷酸酯。称取50 g的PTMEG1000,随后加入多聚磷酸10.2 g,通过剧烈的搅拌混合均匀,随后5次加入5.68 g的粉末状五氧化二磷,保持整个体系温度不超过45 ℃。加料完毕,升温至85 ℃,在此温度下反应6 h后,将体系冷却至室温,加入乙酸乙酯150 mL溶解。用去离子水对有机相进行反复萃取,直至滴加质量分数为10%的氯化钙溶液于水相中无沉淀产生为止,将有机相用无水硫酸镁进行干燥后,旋转蒸发得到透明油状产物PTMEG1000-P,产率为83.7%。1H-NMR (CDCl3, 400 MHz,δ):4.02 (—CH2—O—P,4H),3.35~3.60 (CH2—O—C,64H),1.50~1.75(CH2—C—O,68H);31P-NMR (CDCl3,400 MHz,δ):-9.75 (磷酸二酯,0.17P),-16.00(磷酸单酯,1.0P)
选取分子量为200,400,600和1 000的聚乙二醇PEG,用环氧氯丙烷对其进行环氧化。称取含有0.1 mol羟基的聚乙二醇(PEG200,10 g; PEG400,20 g; PEG600,30 g; PEG1000,50 g),分别加入环氧氯丙烷(27 g,0.3 mol),质量分数91%的氢氧化钠溶液(13.2 g),剧烈搅拌混合均匀后,升温至45 ℃,在此温度下反应2 h后,过滤除去不溶物,滤液用无水硫酸镁进行干燥后,通过减压蒸馏得到淡黄色的黏稠液体PEG-E,(产率:PEG200-E,94.7%; PEG400-E,95.4%; PEG600-E,93.9%; PEG1000-E,93.5%)。1H-NMR (CDCl3,400 MHz,δ):3.65 (m,16.54H (PEG200),34.72H (PEG400),52.90H (PEG600)),3.68(m,2H),3.42 (m,2H),3.11 (m,2H),2.75 (t,2H),2.55 (m,2H)。
2.3以磷酸酯为交联位点的弹性体的制备
在无任何添加剂的情况下,称取定量的不同比例的PTMEG1000-P和不同分子量的PEG-E,与不同含量的GO混合均匀后,浇注到聚四氟乙烯的模具中,在37 ℃下固化,制备磷酸酯交联材料并对其进行简单的命名。其中,以CP表示交联材料仅由PTMEG1000-P和不同分子量的PEG-E混合制备,后面的200,400,600,1 000表示PEG-E原料的分子量,分子量后面的数字表明P-OH与环氧基的物质的量之比(1,2,3,4分别表示P-OH与环氧基的物质的量之比为1∶1,1∶1.5,1∶2,1∶3。CPGO表示交联材料是在CP的基础上添加了GO制备而成,GO后面的百分数表明聚合物中GO的质量分数。例如,CP400-2GO5%表示由PTMEG1000-P,PEG400-E和GO混合制备,其中P-OH与环氧基的物质的量之比为1∶1.5,交联材料中GO的质量分数为5%。
2.4力学性能测试
使用布鲁克公司4302型的万能测试机,根据 ASTM 412-98a 的测试要求对材料进行测试。材料尺寸(26 mm×5 mm×0.7 mm); 拉伸速率:10 mm/min; 所得数据为6次测试数据的平均值。
2.5体外降解测试
交联材料降解性能通过质量的变化来表征。称取交联材料(直径5 mm,高度3 mm)质量记为m0,浸泡于0.1 mol/L、pH = 7.4磷酸盐缓冲溶液(PBS)中。在既定时间内,进行质量测试,记为mt。在降解过程中,随时调整PBS将pH维持在7.4,并保持PBS的体积与实验开始时相同。根据式(1)计算交联材料质量的变化(η),来表征其降解性能。
(1)
2.6红外测试
利用Nicolet 380傅里叶变换红外光谱仪对样品进行红外光谱的检测。由于PTMEG1000-P和PEG-E是黏稠的液体,因此通过涂膜法进行制样检测;由于制备的交联材料具有较好的弹性,利用衰减全反射配件对其进行红外光谱的检测。测试波长为450~4 000 cm-1。
2.7药物释放测试
选取地塞米松作为模拟药物,将50 mg的地塞米松溶解于四氢呋喃(THF)中,加入PEG-E(1 305.5 mg)超声混合后,减压除去THF,加入PTMEG1000-P(695.5 mg)混合均匀,在37 ℃进行固化,即可得到PEG磷酸酯交联的载药体系。复合GO的PEG磷酸酯交联载药体系制备方法同上,将50 mg的地塞米松溶解于THF中,加入PEG200-E(1 305.5 mg)和GO(100 mg),超声溶解混合后,减压除去THF,加入PTMEG1000-P(695.5 mg)混合均匀,在37 ℃进行固化,即可得到复合GO的PEG磷酸酯交联的载药体系。
将上述制备好的载药体系,分隔成100 mg的小块,浸泡于50 mL、0.1 mol/L、pH=7.4的PBS溶液中,放置于37 ℃的恒温培养箱中,在既定时间利用紫外分光光度计检测水相中药物的浓度,收集紫外测试数据,绘制药物释放曲线。
2.8交联材料的体外毒性测试
以PEG200为预聚物的交联材料为例,选择骨髓间质干细胞(BMSCs)为目标细胞,利用MTT比色法对于交联材料进行体外细胞毒性实验。在湿度100%,CO2体积分数为5%,温度37 ℃的条件下,将待测样品浸泡于改良杜氏伊格尔(DMEM)培养基中。24 h后,取上层清液过滤并杀菌(样品浸液)。将已在DMEM培养基中培养4 h的BMSCs(每孔5×103个)中的培养基用上述样品浸液替代,然后继续分别培养24 h和48 h,随后加入二甲亚矾(DMSO)终止反应。通过酶联检测仪在490 nm下分别测试了BMSCS在样品浸液中培养24 h和48 h的吸光度值(Atest),BMSCS在培养基中培养相同时间的吸光度值(Acontrol)以及空白DMEM培养基的吸光度值(A0)材料的检测吸光度值。细胞相对毒性计算如下(RCV):
(2)
3 实验结果及讨论
3.1磷酸化PTMEG1000和环氧化PEG结构的分析和表征
多聚磷酸和五氧化二磷的混合磷酸化试剂因产物磷酸化高,P原子的利用率高,产物中磷酸单酯含量高等优异性[20]被应用为本实验中的磷酸化试剂。如图1(a)所示,在1H-NMR中,代表PTMEG1000—CH2—OH,δ为3.50~3.60的信号峰完全消失;出现δ在4.02的—CH2OP(O)的信号峰,表明PTMEG1000中的羟基已经完全转换为磷酸酯的形式。疏水性的磷酸酯与六甲基二硅氮烷反应后形成硅化的磷酸酯,在磷谱中具有不同的化学位移[21],硅化后磷酸单酯的δ在-17左右,硅化后磷酸双酯的δ在-10左右,通过积分可以换算出各组分的物质的量之比。根据此方法,硅化处理PTMEG1000-P,其31P-NMR如图1(b)所示,δ在-16.04处为磷酸单酯,化学位移在-10.30为磷酸二酯,通过积分换算,PTMEG1000-P中单双酯的物质的量之比为100∶13。证明PTMEG1000-P被成功制备,且具备较高的磷酸单酯含量。
本实验中采用不同分子量的PEG与环氧氯丙烷和高浓度的氢氧化钠溶液反应来制备PEG-E。与其他方法比较,在环氧化过程中未添加具有毒性的相转移催化剂[22],而是利用PEG本身的相转移催化作用[23]进行PEG-E的制备。如图2所示,δ为2.55、2.75处的信号峰对应含氧杂环中—CH2—的H,δ为3.11处的信号峰对应含氧杂环中—CH—中的H,δ为3.42、3.68处的信号峰对应含氧杂环与PEG主链相连接处—CH2—中的H,证实了PEG-E中缩水甘油醚结构的存在,即PEG-E被成功制备。

图1 PTMEG1000-P的1H-NMR谱图(a)和31P-NMR谱图(b)Fig.1 1H-NMR spectra (a) and 31P-NMR spectra of PTMEG1000-P (b)
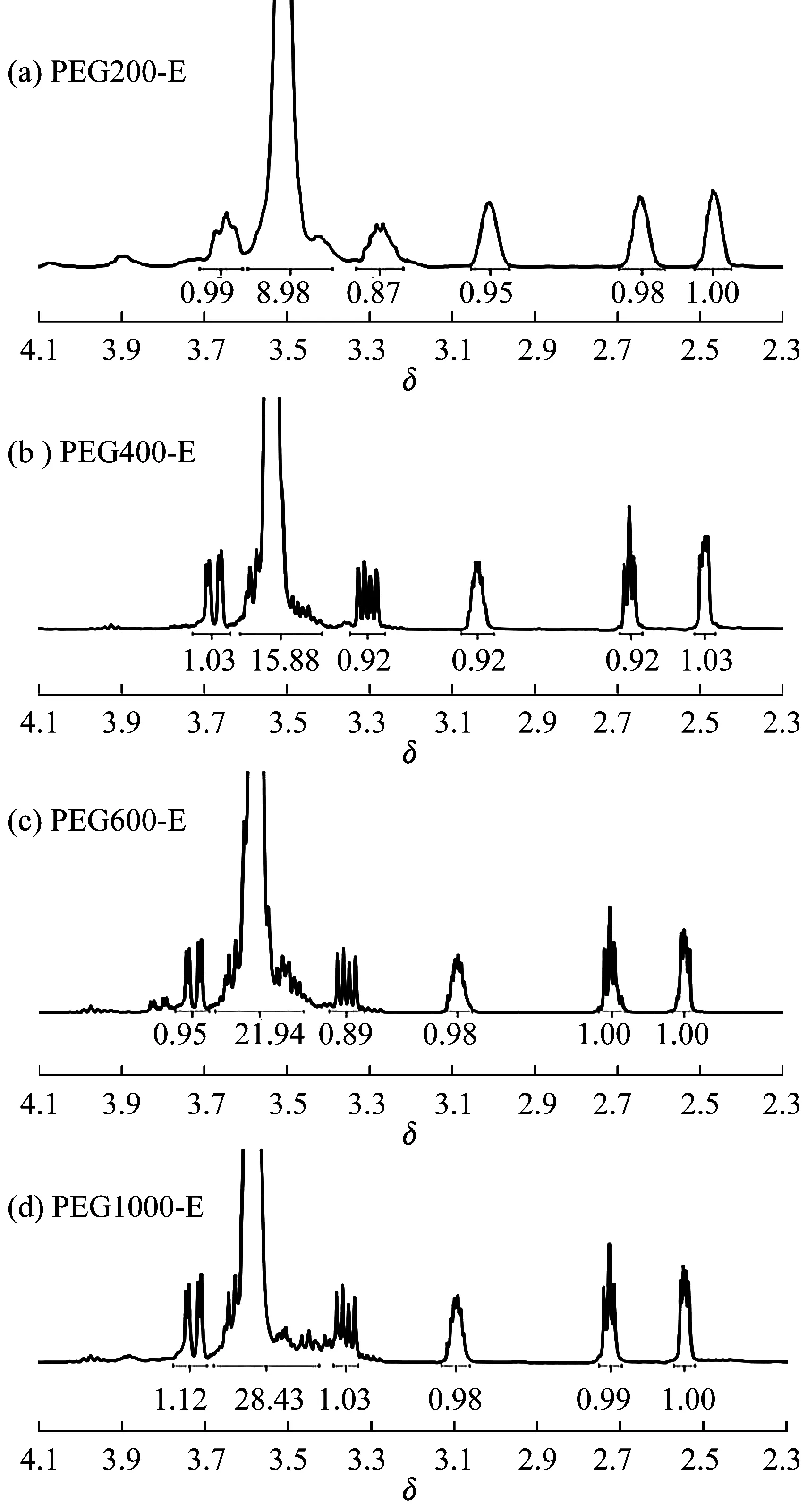
图 2 PEG-E的 1H-NMR谱图Fig.2 1H-NMR spectra of PEG-E
3.2力学性能测试
交联材料的力学性能如图3所示。图3(a)示出了未添加GO的交联弹性体的杨氏模量,在预聚物比例相同时,随着PEG-E的分子量的升高,交联材料的杨氏模量减小,说明交联材料的交联密度随着预聚物分子量的升高而减少,其中PEG1000-E与PTMEG1000-P的混合物由于交联密度过小没有形成交联弹性体; 在确定反应原料后,随着体系中环氧基团添加量的增加,交联材料的杨氏模量先升高后降低,其中在P-OH与环氧基的物质的量之比为1∶1.5时杨氏模量最大,表明此比例为本体系最佳的交联配比,其交联密度最大。当选取P-OH与环氧基的物质的量之比为1∶1.5时,图3(b)所示的是不同GO添加量对交联材料的杨氏模量的影响。GO的添加使得材料的杨氏模量得到提高,并随着GO添加量的增加,交联材料的杨氏模量随之升高。

图3 交联材料的力学性能测试Fig.3 Mechanical test for crosslinked polymers
对PEG-E进行弹性测试是必须的。图3(c)所示的是PEG-E不同分子量和P-OH与环氧基不同比例对交联材料弹性的影响。从图中可以看出制备的几种交联弹性体的断裂伸长率均大于100%,证明材料具有较好的弹性。随着预聚物分子量的升高,材料的断裂伸长率也随之升高。在固定原料后,材料的断裂伸长率随着体系中环氧基团含量的提高,先降低后升高,P-OH与环氧基的物质的量之比为1∶1.5时断裂伸长率最小。这说明断裂伸长率也是和材料的交联密度密切相关的,随着交联密度的提高,材料的断裂伸长率下降,结合图2(a)分析,制备的交联弹性体的断裂伸长率和杨氏模量成反比。图4(d)所示的是GO的添加降低了材料的断裂伸长率,并且断裂伸长率随着GO含量的增加而降低,这可能是由于GO的加入破坏了材料内部的规整性导致的。
因此可以通过调节预聚物的分子量、比例和GO的添加量来制备不同力学性能的交联弹性体。
3.3体外降解实验
图4所示的是材料的体外降解实验。结果表明,经过一开始的吸收增重,到后面的逐步降解,制备的各种材料在经历不同的时间后均可以完全降解。图4(a)中表明,CP200-2的降解时间最长,这说明材料的降解性能和交联密度具有一定关系。图4(b)中,随着原料分子量的增加交联材料降解时间减少,也补充说明了降解性能受交联密度的影响,交联密度大,降解时间长。GO表面具有大量的含氧基团,可以增加材料的亲水性,同时含有的羧酸基团对交联位点磷酸酯的水解起到一定的催化作用,同时GO的加入破坏了材料内部的规整性,也会加快材料的降解。图4(c)所示,GO的加入确实减少了材料的降解时间,但是随着GO添加量的改变,材料的降解时间变化不大。
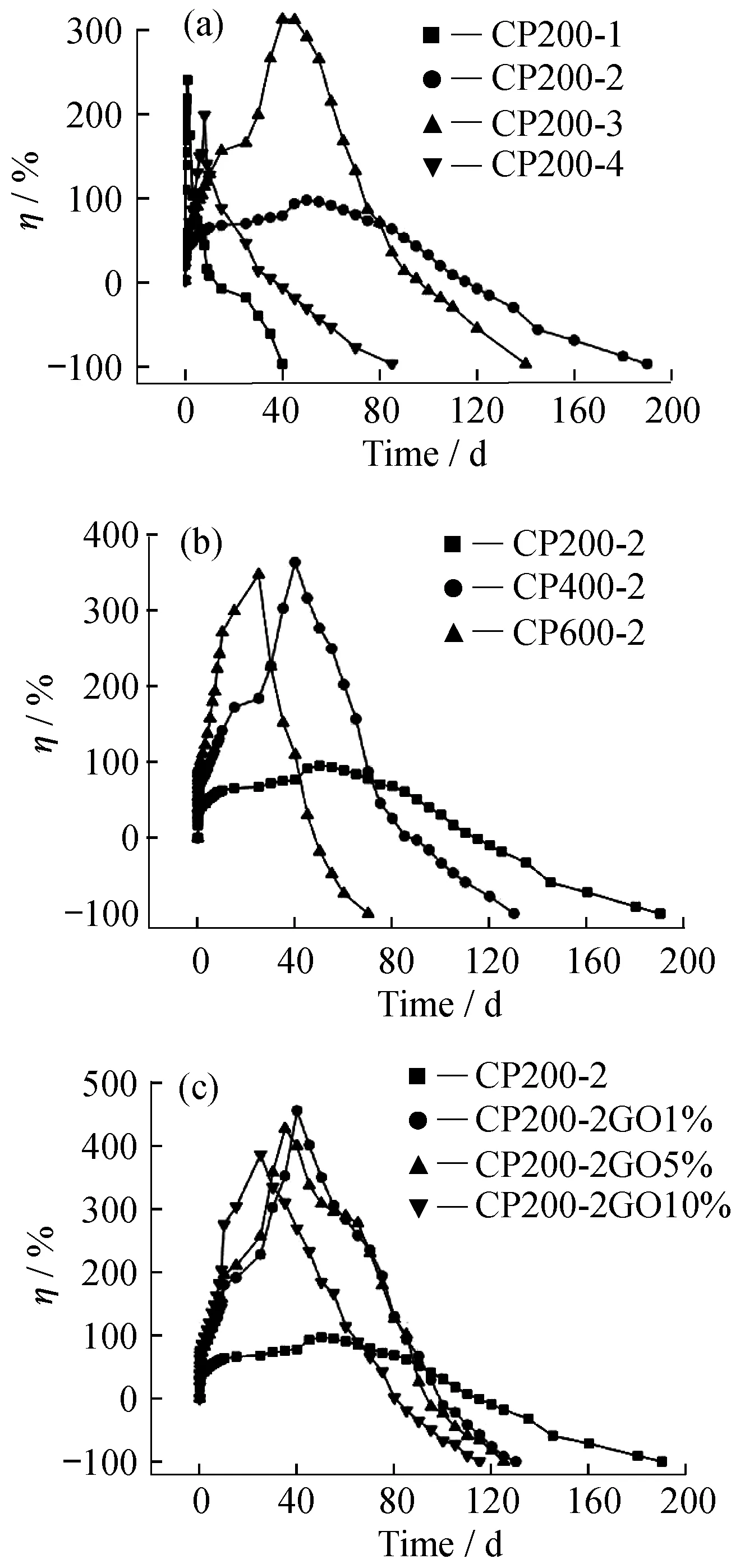
图4 交联材料的降解性能测试Fig.4 Degradability test for crosslinked polymers
上述结果证明本实验中制备的交联弹性体具备完全降解性,并且通过改变原来的分子量、比例可以调节材料的降解性能,同时GO的添加也可以促进材料的降解。
3.4红外测试研究
理论上,当各组分恰好完全反应时,制备的交联材料的交联密度会达到最大。本实验中,P-OH与环氧基是等物质的量反应,理论上当P-OH与环氧基的物质的量之比为1∶1时,材料获得最大的交联密度。通过对材料力学性能和体外降解性能的分析,制备的交联弹性体在P-OH与环氧基的物质的量之比为1∶1.5时获得最大的交联密度。如图5(a)所示,通过红外测试交联后的材料与原料相比,并没有出现明显的特征吸收,只是在909 cm-1环氧基团特征吸收峰处[24]发生了信号变化。在CP200-2中,P-OH与环氧基团物质的量之比为1∶1.5,环氧基团过量,但是并未发现909 cm-1处的环氧基团特征吸收峰,甚至在3倍的环氧基团加入后也几乎没有发现环氧基团的特征吸收峰。根据Penczek[25]的研究,推测在本实验体系中除了P-OH与环氧基团发生开环反应以外,还存在消耗环氧的其他基团。P-OH首先与环氧基团发生开环反应,同时生成了C-OH,随后,在P-OH与环氧基团继续反应生成磷酸酯交联位点的同时,前一步生成的C-OH也会与环氧基团发生开环反应。图5(b)所示的是加入了GO的交联弹性体的红外谱图,在1 732 cm-1和1 642 cm-1的特征吸收峰,分别对应C=O的伸缩振动和未被完全氧化的C=C的伸缩振动,表明交联材料中GO的存在,GO中1 225 cm-1处的C-OH的特征吸收峰和3 430 cm-1附近的—OH伸缩振动峰未被发现,可能是由于GO浓度较低,被磷酸酯交联材料所包裹导致的。通过与未添加GO的交联弹性体红外谱图比较,未发现其他新的吸收峰。结合交联材料的力学性能的表征,推断含有GO的交联材料的形成机理与未含GO的交联材料相同,如图6所示,GO通过氢键以及物理吸附作用与磷酸酯交联材料连接在一起,作为一种无机填料存在于交联体系中。

图5 交联材料的红外谱图Fig.5 IR spectra of crosslinked polymers
3.5药物释放研究
选取地塞米松为模拟药物,通过简单的混合搭载到交联材料上,其药物释放曲线如图7所示。未添加GO的交联材料的药物释放主要依靠可降解高分子在吸水溶胀过程中,药物溶解于水中来实现。在药物释放前期,释放速率很快,10 h就释放了载药量的60%左右,随着溶液中药物浓度的升高,释放速率逐渐变慢,最终达到释放平衡。以PEG400-E和PEG600-E为原料制备的交联材料CP400-2和CP600-2的药物释放体系在48 h基本释放完全,而以低分子PEG200-E为原料的CP200-2在120 h左右才将药物完全释放,这也与前文对交联材料降解性能的表征结果一致。随着原料分子量的提高,交联材料的交联密度减少,材料的降解速率提高,因此可以通过调整原料的分子量、比例等来控制交联材料的交联密度从而满足不同需求的药物释放。对于添加了GO的交联材料,虽然其降解速率要比相应的未添加GO的交联材料快,但是通过分析CP200-2和CP200-2GO5%的药物释放曲线可知,CP200-2GO5%的药物释放速率要小于CP200-2。我们推测,这是由于模拟药物通过氢键和π-π作用与GO进行结合,延缓了药物的释放,起到了一定的药物缓释作用。

图6 PTMEG1000-P,PEG-E和GO形成交联结构示意图Fig.6 Schematic presentation for the formation crosslinked polymers from PTMEG1000-P,PEG-E and GO
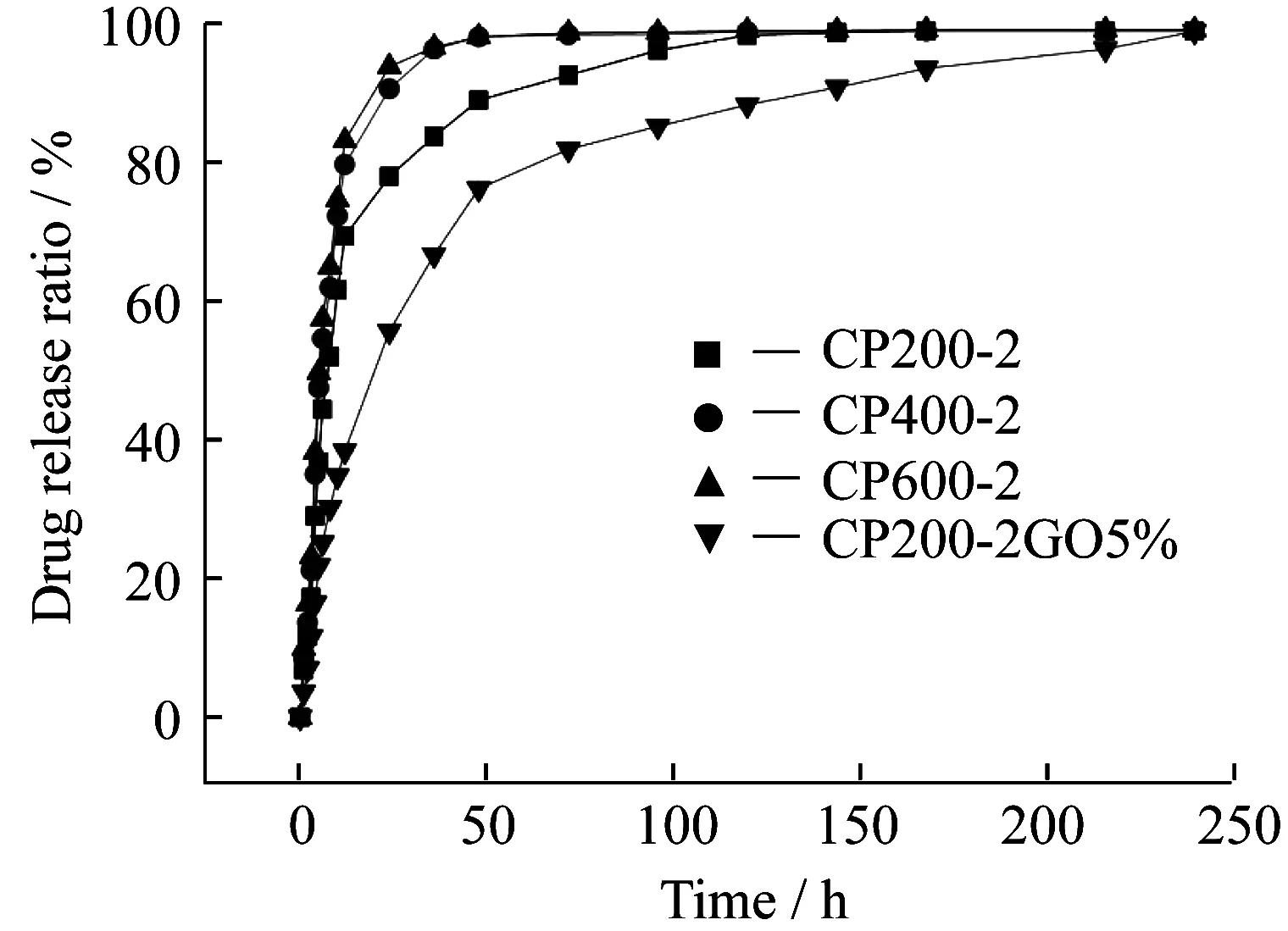
图7 交联材料在pH=7.4的PBS溶液中的药物释放曲线Fig.7 Drug release curve from drug contained CPs in PBS solution(pH=7.4)
3.6体外细胞毒性测试
对生物体无毒是对生物材料最基本应用要求,因此对生物材料的毒性测试是必需的。我们选择了具有较好药物缓释作用的CP200系列交联材料进行体外细胞毒性测试。如图8所示,在未添加GO的交联材料中,CP200-1的细胞相对活性最低,这是由于存在于反应的P-OH导致培养液的pH值偏低,影响了细胞的增殖,但是其存活率在80%以上,基本可以满足生物应用。随着环氧含量的提高,CP200-2,CP200-3和CP200-4的细胞相对存活率均在85%以上,具有较低的细胞毒性。添加了GO的交联材料,以CP200-2GO5%为例,其细胞相对存活率超过了95%,证明GO的添加可以提高交联材料的细胞相容性。

图8 交联材料对BMSCs的体外细胞毒性测试Fig.8 Cell viability of BMSCs with CPs in vitro
4 结 论
本文在未添加相转移催化剂的条件下制备了不同分子量的环氧化聚乙二醇(PEG-E),在无溶剂条件下通过与磷酸化聚四氢呋喃二醇(PTMEG-P)和GO的简单物理混合制备了一系列的交联材料。通过实验测试可知,改变原料的分子量、比例以及GO的添加量可以控制交联材料的力学性能、降解性能和药物释放性能以满足不同的需求。体外细胞毒性测试表明,制备的交联材料具有较低的细胞毒性,在生物医学领域具有一定的应用前景。
[1]FENG Liangzhu,LIU Zhu.Graphene in biomedicine:Opportunities and challenges[J].Nanomedicine,2011,6(2):317-324.
[2]KOVTYUKHOVA N I,OLLIVIER P J,MARTIN B R,etal.Layer-by-layer assembly of ultrathin composite films from micron-sized graphite oxide sheets and polycations[J].Chemistry of Materials,1999,11(3):771-778.
[3]KAI Weihua,HIROTA Y,HUA Lei,etal.Thermal and mechanical properties of a poly(ε-caprolactone)/graphite oxide composite[J].Journal of Applied Polymer Science,2008,107(3):1395-1400.
[4]SUN Xiaoming,LIU Zhuang,WELSHER K,etal.Nano-graphene oxide for cellular imaging and drug delivery[J].Nano Research,2008,1(3):203-212.
[5]LIU Zhuang,ROBINSON J T,SUN Xiaoming,etal.Pegylated nanographene oxide for delivery of water-insoluble cancer drugs[J].Journal of the American Chemical Society,2008,130(33):10876-10877.
[6]ZHANG Liming,XIA Jingguang,ZHAO Qinhuan,etal.Functional graphene oxide as a nanocarrier for controlled loading and targeted delivery of mixed anticancer drugs[J].Small,2010,6(4):537-544.
[7]LI Shanghao,APHALE A N,MACWAN I G,etal.Graphene oxide as a quencher for fluorescent assay of amino acids,peptides,and proteins[J].Applied Materials & Interfaces,2012,4(12):7069-7075.
[8]ZHANG Wen,GUO Zhouyi,HUANG Deqiu,etal.Synergistic effect of chemo-photothermal therapy using PEGylated graphene oxide[J].Biomaterials,2011,32(33):8555-8561.
[9]杨永岗,陈成猛,温月芳.氧化石墨及其与聚合物的复合[J].新型炭材料,2008,23(3):193-200
[10]BARROSO-BUJANS F,CERVENY S,VERDEJO R,etal.Permanent adsorption of organic solvents in graphite oxide and its effect on the thermal exfoliation[J].Carbon,2010,489(4):1079-1087.
[11]SHIKANOV A,SMITH R M,XU Min,etal.Hydrogel network design using multifunctional macromers to coordinate tissue maturation in ovarian follicle culture[J].Biomaterials,2011,32(10):2524-2531.
[12]PENG Ke,TOMATSU I,KOROBKO A V,etal.Cyclodextrin-dextran basedinsituhydrogel formation:A carrier for hydrophobic drugs[J].Soft Matter,2010,6(1):85-87.
[13]ZUSTIAK S P,LEACH J B.Hydrolytically degradable poly(ethylene glycol) hydrogel scaffolds with tunable degradation and mechanical properties[J].Biomacromolecules,2010,11(5):1348-1357.
[14]史洋,魏东芝.用活化的单甲氧基聚乙二醇修饰L-天冬酰胺酶[J].华东理工大学学报(自然科学版),2001,27(6):601-604.
[15]WEBER L M,LOPEZ C G,ANSETH K S.Effects of PEG hydrogel crosslinking density on protein diffusion and encapsulated islet survival and function[J].Journal of Biomedical Materials Research:Part A,2009,90 A(3):720-729.
[16]LIN C C,ANSETH K S.PEG Hydrogels for the controlled release of biomolecules in regenerative medicine[J].Pharmaceutical Research,2009,26(3):631-643.
[17]LAPIENIS G,PENCZEK S.Kinetics and thermodynamics of the polymerization of the cyclic phosphate esters:II.Cationic polymerization of 2-methoxy-2-oxo-1,3,2-dioxaphosphorinane (1,3-propylene methyl phosphate)[J].Macromolecules,1974,7(2):166-174.
[18]BIELA T,KUBISA P.Oligomerization of oxiranes in the presence of phosphoric acid:Kinetics of model reaction[J].Die Makromolekulare Chemie,1991,192(3):473-489.
[19]HUMMERS W,Offeman R E.Preparation of graphitic oxide[J].Journal of the American Chemical Society,1958,80(6):1339-1339.
[20]TRACY D J,REIERSON R L.Commercial synthesis of monoalkyl phosphates[J].Journal of Surfactants and Detergents,2002,5(2):169-172.
[21]PENCZEK S,KALUZYNSKI K,PRETULA J.Addition of H3PO4to diglycidyl ethers of bisphenol A:Kinetics and product structure[J].Journal of Applied Polymer Science,2007,105(1):246-254.
[22]CRIVELLO J V.Design and synthesis of multifunctional glycidyl ethers that undergo frontal polymerization[J].Journal of Polymer Science:Part A.Polymer Chemistry,2006,44(21):6435-6448.
[23]张新胜,祁国珍.相转移催化反应的研究:Ⅳ.聚乙二醇催化机理[J].华东理工大学学报(自然科学版),1994,20(6):782-787.
[24]LAN Xiaokang,HUANG Wei,YU Yuzhao.Synthesis,characterization and properties of the polysiloxane-based episulfide resin[J].European Polymer Journal,2010,46(7):1545-1556.
[25]PENCZEK S,KALUZYNSKI K,PRETULA J.Phosphorylation of polyols with H3PO4:Towards simple synthesis of poly(alkylene phosphate)s[J].Phosphorus,Sulfur and Silicon,2009,184(8):1935-1945.
Phosphoester Cross-Linked Elastomer with Graphene Oxide:Synthesis and Application as Drug Carrier
SUN Huang-hui1,XU Zhi-zhen1,XIA Wei1,ZHANG Shu1,ZHANG Li-wei2,ZHANG Wen-qing1
(1.Shanghai Key Laboratory of Functional Materials Chemistry,East China University of Science and Technology, Shanghai 200237,China;2.Yunnan Linyuan Perfume Co.Ltd,Yuxi 651100,Yunnan,China)
Based on a facile crosslinked reaction between diphosphoesters of polytetramethylene-oxide glycol (PTMEG1000-P) and diglycidyl ether of polyethylene glycol (PEG-E) prepared without any phase-transfer catalyst with graphene oxide (GO),a new series of degradable crosslinked phosphoester polymers (CPs) were prepared and characterized.The relevant experiments suggested good mechanical and degradable properties of the polymers.GO played an important role for the properties of CPs polymers,such as mechanical property and degradation rate.Drug release tests and biological cytotoxicity studiesinvitrosuggested that the polymers had great potential applications in drug delivery systems.
graphene oxide; phosphate ester bond; biomaterials; drug carrier
1006-3080(2016)04-0513-08
10.14135/j.cnki.1006-3080.2016.04.012
2016-03-28
云科合发(2014)5号; 云南合作院士工作站资助
孙黄辉,男,山东烟台人,博士生,研究方向为生物材料。E-mail:apll0105@live.cn
通信联系人:张文清,E-mail:zhwqing @ecust.edu.cn
R318.08
A
