拉压变形对B(N)掺杂碳纳米管Al吸附性能的影响*
2016-09-14刘贵立宋媛媛王天爽
刘贵立, 宋媛媛, 姜 艳, 周 爽, 王天爽
(沈阳工业大学 建筑与土木工程学院, 沈阳 110870)
拉压变形对B(N)掺杂碳纳米管Al吸附性能的影响*
刘贵立, 宋媛媛, 姜艳, 周爽, 王天爽
(沈阳工业大学 建筑与土木工程学院, 沈阳 110870)
为了研究(5,5)碳纳米管的B(N)环状掺杂效应,以及拉压变形对B(N)环状掺杂碳纳米管Al吸附性能的影响,采用基于密度泛函理论的平面波赝势和广义梯度近似方法,对B(N)环状掺杂碳纳米管Al吸附模型进行了几何优化,计算了B(N)环状掺杂碳纳米管的形成能,并确定了Al原子的最稳定吸附位置.结果表明,B(N)环状掺杂(5,5)碳纳米管的结构较为稳定.B(N)环状掺杂可以增加碳纳米管与Al之间的吸附能;一定范围内的拉伸和压缩变形则会降低碳纳米管与Al之间的大部分吸附能.
碳纳米管; 第一性原理; 掺杂; Al原子吸附; 拉压变形; 吸附能; 密度泛函理论; 几何优化
碳纳米管(CNT)是一种具有特殊结构的一维量子材料,具有许多特殊力学、电学和化学性能,在纳米电子器件[1]、纳米机械复合增强材料[2-7]及储氢材料[8-9]等众多领域取得了广泛应用.在碳纳米管增强复合材料方面,闫瑞芳等[10]采用挤压铸造法成功制备了CNTs混杂增强2024Al复合材料;邓春锋等[11]采用冷等静压热挤压方法制备了碳纳米管增强2024Al基复合材料,通过CNT表面改性(如包覆、缺陷和掺杂等)可以改善CNT分散性差且与金属基体结合较弱等问题.目前对碳纳米管和石墨烯纳米带的掺杂主要集中在B(N)掺杂上,徐慧等[12]研究了B/N共对掺杂碳纳米管;刘贵立等[13]用第一性原理研究了掺杂N,B和Si的超晶格电子结构.
在CNT与金属模拟方面,唐玉琴[14]采用批式法分别研究了Eu和Cu在多壁碳纳米管上的吸附行为;张变霞[15]采用密度泛函方法对铜原子在有限长(5,5)椅型碳纳米管的吸附行为进行了研究;张国英等[16]在介观尺度研究了CNT增强Al晶粒的电子结构.本文基于密度泛函理论的第一性原理,计算了B(N)原子环状掺杂的形成能,还对环状掺杂B和N的(5,5)碳纳米管不同位置上Al原子的吸附在无变形和拉压变形下进行了几何优化以及能量计算.
1 理论模型与计算方法
本文基于密度泛函理论,利用软件Acclerys Material Studio中的CASTEP模块对吸附金属Al原子前后的(5,5)CNT体系进行结构优化和总能量计算.纯CNT、掺杂CNT以及Al原子吸附的模型如图1所示.由于金属Al晶体中Al原子间距最小为0.286 nm,为了避免Al原子之间的作用对吸附能计算的影响,采用两个重复单元为一周期,其中Al间最小距离为0.492 nm,符合要求.
计算交换关联势采用广义梯度近似(GGA)修正的PW91泛函,价电子波函数的截止能量设置为340 eV,几何优化过程中收敛精度为1.0×10-6eV,作用在每个原子上的压力不大于0.03 eV/A,晶体内应力不大于0.05 GPa,布里渊区K点设置为1×1×10.
拉伸和压缩模型是在图1c模型的基础上,固定底层原子,对第二至四层原子分别均匀施加轴向拉伸或压缩位移荷载,对不同Al吸附位置的模型采用第一性原理计算软件Acclerys Material Studio中CASTEP模块进行几何优化和能量计算.本文拉伸总变形分别取0.015、0.03、0.045和0.09 nm;压缩总变形分别取0.015、0.03、0.045和0.09 nm.
2 结果分析与讨论
2.1硼氮环状掺杂对(5,5)CNT的影响
为了分析B(N)环状掺杂对碳纳米管稳定性的影响,计算了(5,5)掺杂碳纳米管的总能量,并计算了B(N)环状掺杂碳纳米管的形成能,形成能表示为
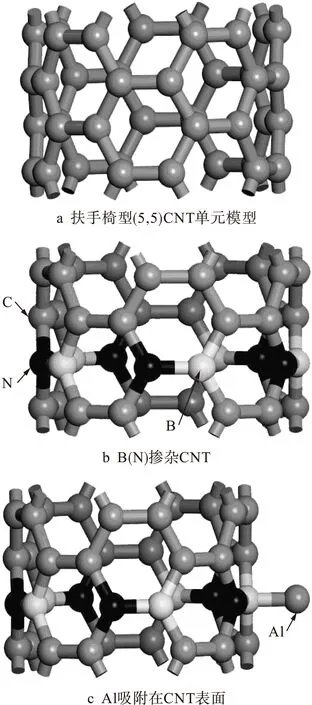
图1 碳纳米管模型Fig.1 Model for CNT
(1)
式中:Et1为碳纳米管掺杂或者引入缺陷后的总能量;Et2为未掺杂碳纳米管的总能量;Ex为掺杂B原子的单个原子能量;Ey为掺杂N原子的单个原子能量;l为晶胞的总原子数;m为掺杂B原子数;n为掺杂N原子数.m与n均为5,计算过程中硼氮原子的能量是不变的,分别是-70.490 5 eV和-262.696 3 eV.计算得出硼氮环状掺杂碳纳米管的形成能为-79.384 315 eV,说明硼氮环状掺杂是能量减少的过程,需要放出能量.
B(N)环状掺杂(5,5)CNT优化结构后,B—C键增大约4.2%,N—C减小0.5%,B—N键增大2.6%,C—C键减小0.99%,键长和键角发生了一些微小变化,但掺杂B、N不影响CNT石墨壁六元环结构.
2.2掺杂Al对CNT表面吸附结构的影响
首先在吸附前对(5,5)CNT进行优化,在整个过程中体系能量呈下降趋势,最终收敛构型中C—C键长约在0.141 5~0.142 nm之间,与文献[17]中的C—C键长在0.142 2~0.142 7 nm之间很接近,最大误差仅为0.8%,证明计算结果是可靠的.(5,5)CNT模型中Al原子可能吸附于CNT外壁的初始位置有16种,如图2所示.
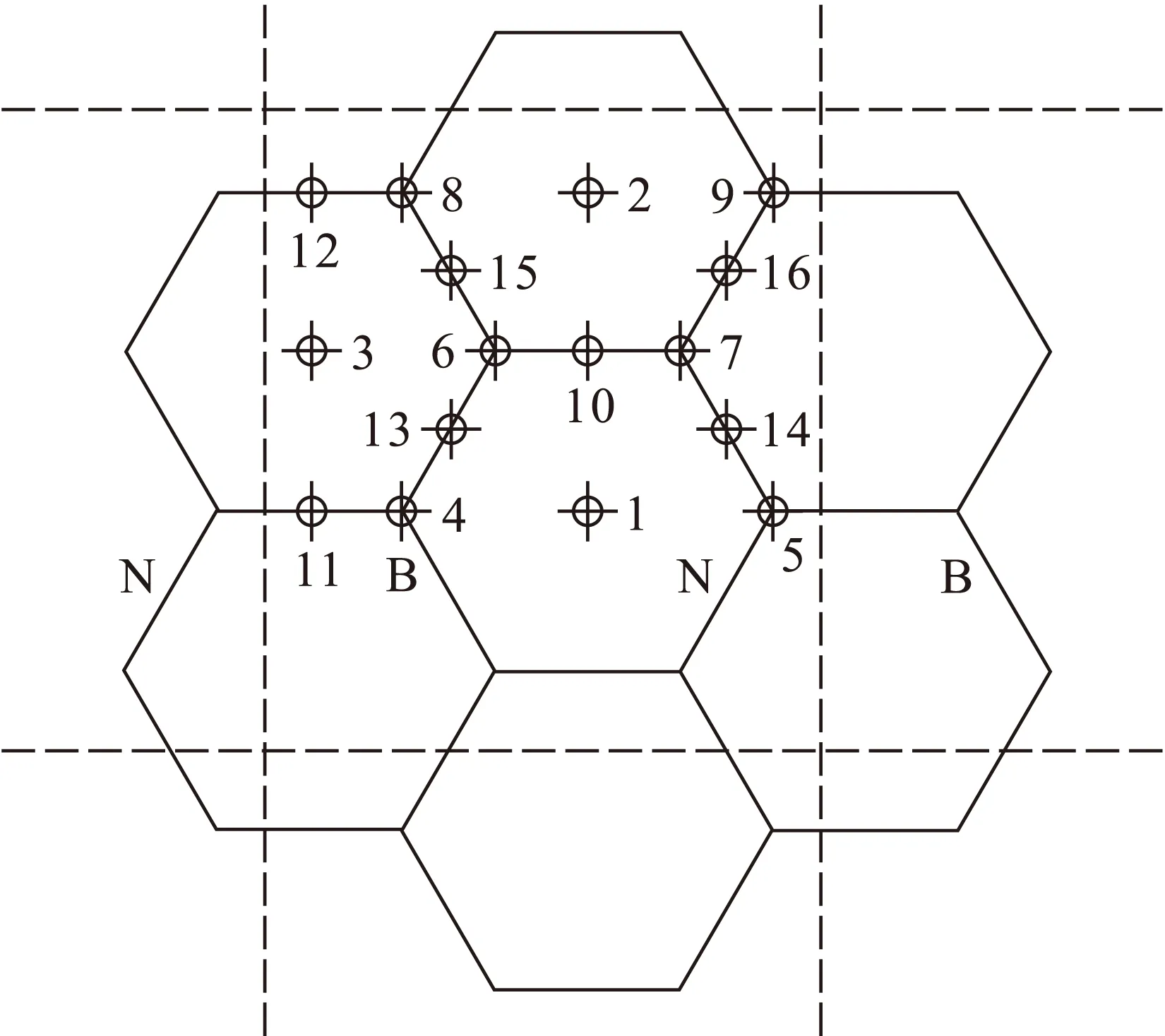
图2 Al原子的初始吸附位置Fig.2 Initial adsorption positions of Al atoms
图2中虚线内为最小循环周期,1,2,3位于石墨六边形单元中心上方,4,5,6,7,8,9位于原子上方,10,11,12,13,14,15,16位于C—C键、C—N键、C—B键或N—B键中心上方.Al—C初始键长设为0.188 nm,Al—B和Al—N键长初值分别设定为0.182和0.185 nm.
原子吸附能Ea定义为

(2)
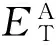
通过对吸附Al的CNT体系优化获得总能量,并计算得到的吸附能为1.24~1.42 eV,而吸附Al的B(N)环状掺杂(5,5)CNT的16种位置的吸附能计算结果如表1所示.
除10号初始位置外吸附能均大于0 eV,反应可以进行.根据吸附能大小及Al原子最终收敛位置可以将初始吸附位置分为三类,如图3所示.
吸附能较大的点2,3,6,8,12,15为A类,最终稳定位置为图中A范围,偏离中心0.019 4 nm,B(N)的掺杂使(5,5)CNT对Al原子的吸附能有所提高;吸附能较小的点5,7,9,14,16为B类,最终稳定位置为图中B范围,偏离中心0.026 06 nm,B(N)的掺杂使(5,5)CNT对Al原子的吸附能基本没有提高;吸附能再小的点1,4,11,13为C类,最终稳定位置为图中C范围,偏离中心0.015 24 nm,B(N)的掺杂使(5,5)CNT对Al原子的吸附能降低.吸附能最高的A类原子位置最终稳定在距离B(N)掺杂环较远的碳环中心偏向B原子的上方,Al在A范围形成具有较高吸附能的稳定结构,在B,C范围形成具有较低吸附能的亚稳定结构.总之,从吸附前后的吸附能及几何构型来看,环状掺杂B(N)有助于提高Al在CNT外壁的吸附能.
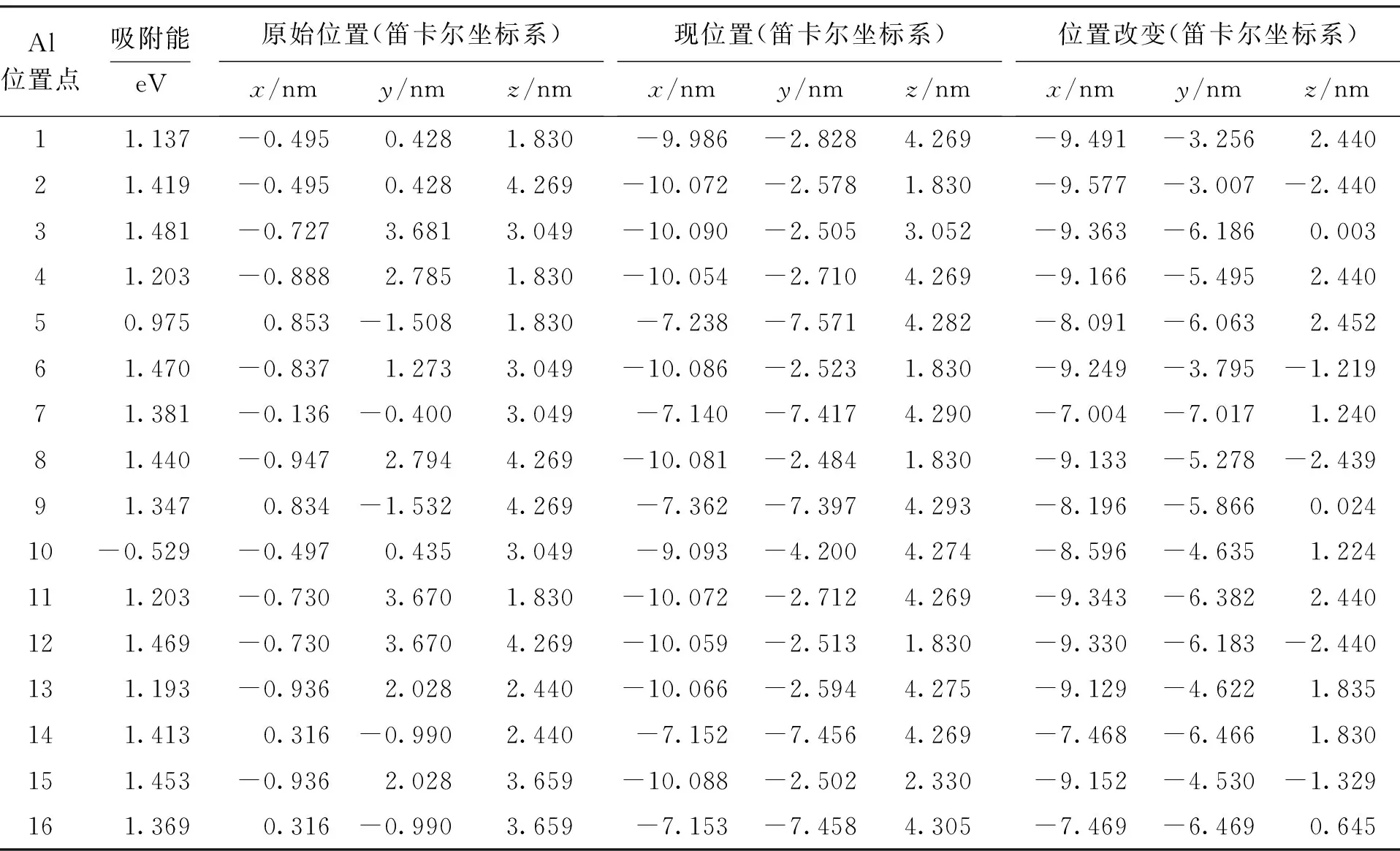
表1 不同Al初始位置的吸附能Tab.1 Adsorption energy at different Al initial positions
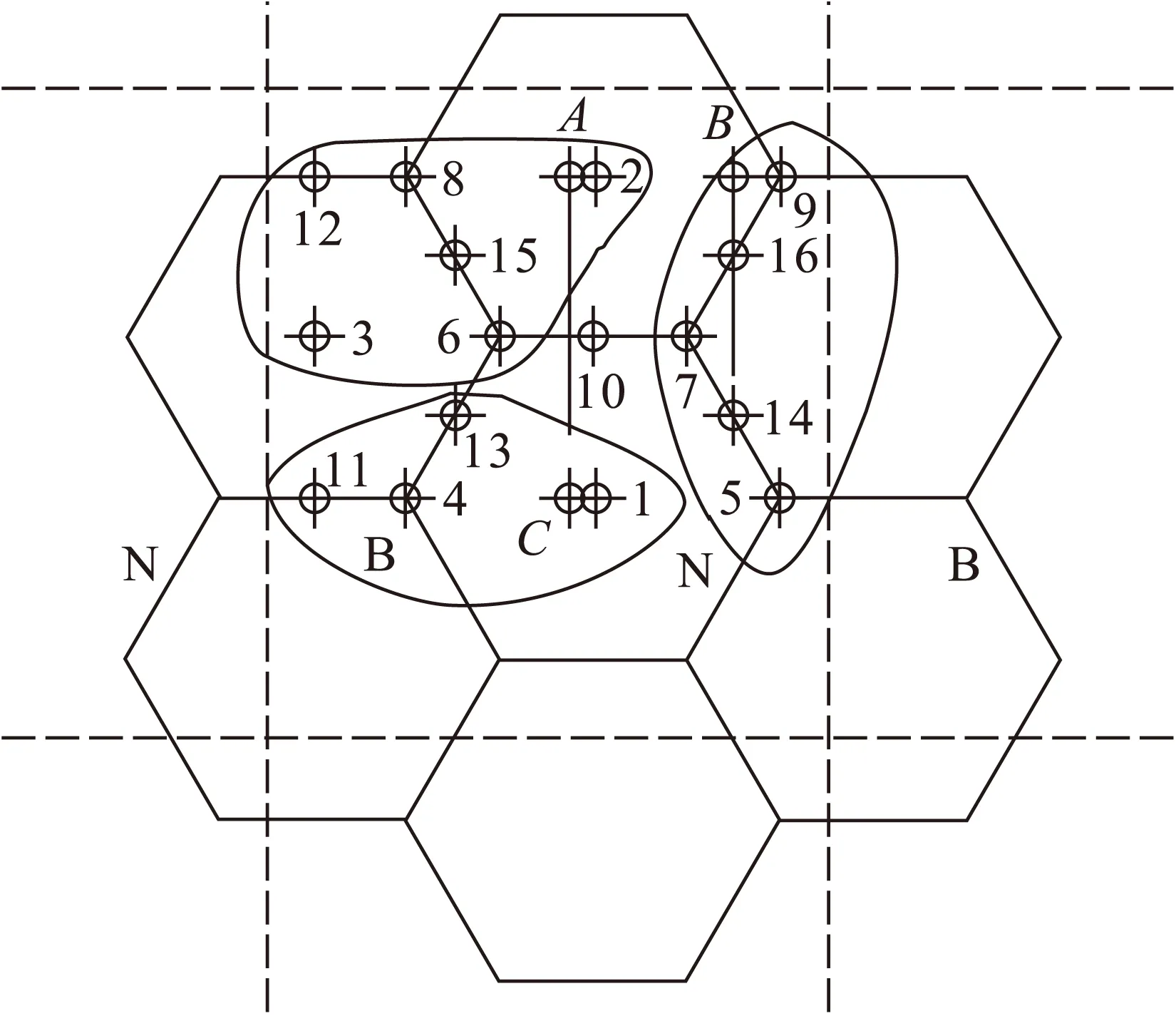
图3 Al原子的稳定吸附位置分类Fig.3 Stable adsorption positions of Al atoms
2.3拉伸和压缩对Al在掺杂CNT表面的影响
基于建立的拉伸和压缩模型,选取部分A,B,C三类吸附能较大的Al原子吸附位置进行结构优化和能量计算,计算结果如表2~4所示.
A类和B类初始Al吸附位置的模型在拉伸(5,5)CNT 0.015~0.09 nm的情况下,吸附能均减小,并且Al原子在CNT外壁最终稳定位置基本不变.其中B类初始Al吸附位置吸附能减小0.31~0.35 eV,吸附能减小约23%~25%;A类初始Al吸附位置吸附能减小0.26~0.27 eV,吸附能减小大约17%~19%;C类初始Al吸附位置模型在拉伸(5,5)CNT 0.015~0.03 nm时,吸附能减小0.25~0.26 eV,拉伸到0.045 nm时,位置13处吸附能基本不变,拉伸到0.09 nm时,C类初始Al吸附位置处的吸附能均基本不变,并且所有原子的最终稳定位置与A类相同.综上所述,对B(N)环状掺杂(5,5)CNT一定范围内的拉伸会引起吸附能较强的吸附位置Al原子在掺杂CNT表面吸附能降低,并且稳定位置基本不变;而在吸附能较弱的吸附位置的Al原子在掺杂CNT表面吸附能有所提高,并且引起稳定位置向强吸附能的位置转移.
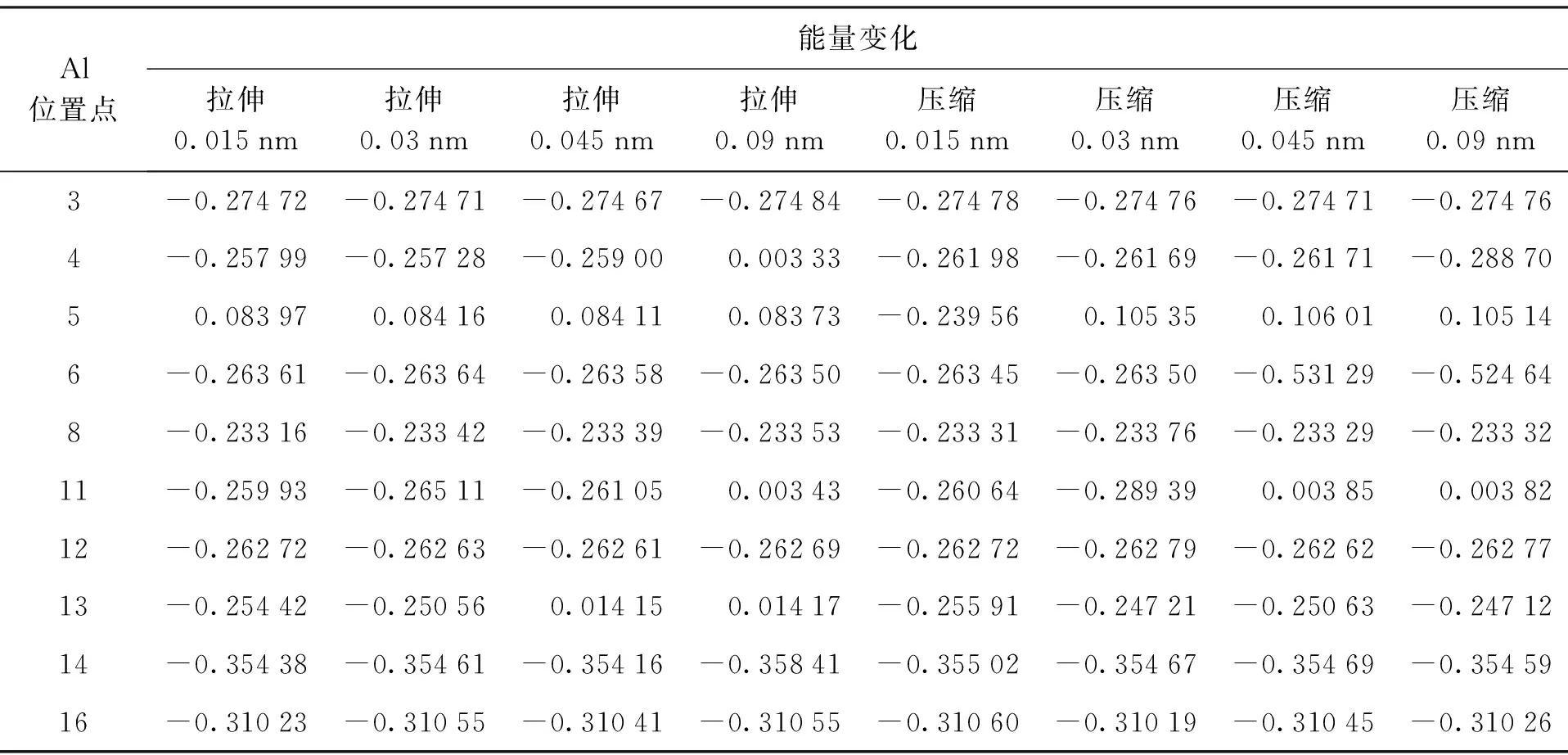
表2 不同拉伸和压缩长度下的吸附能Tab.2 Adsorption energy under different tensile and compression lengths eV
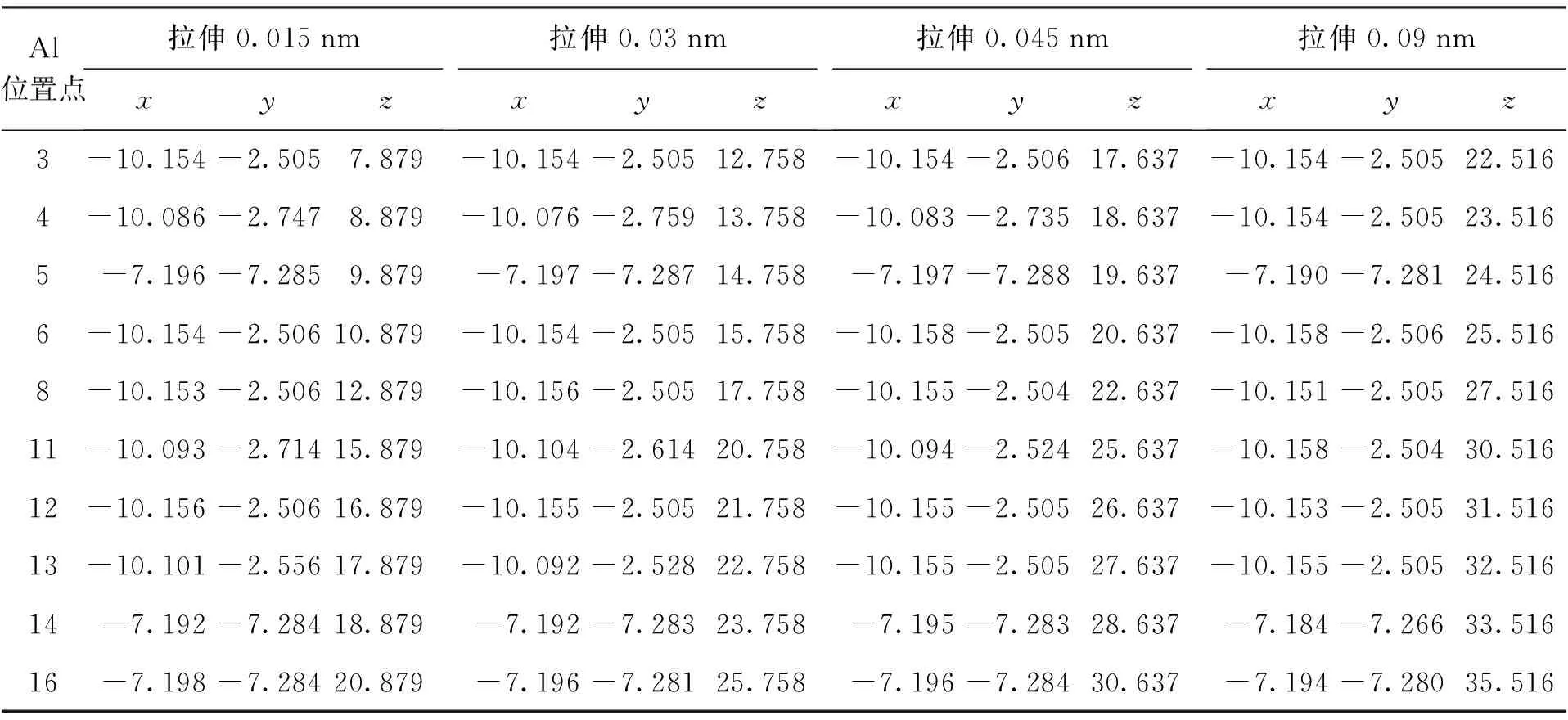
表3 不同拉伸长度下Al的最终稳定吸附位置Tab.3 Final stable adsorption positions of Al under different tensile lengths nm
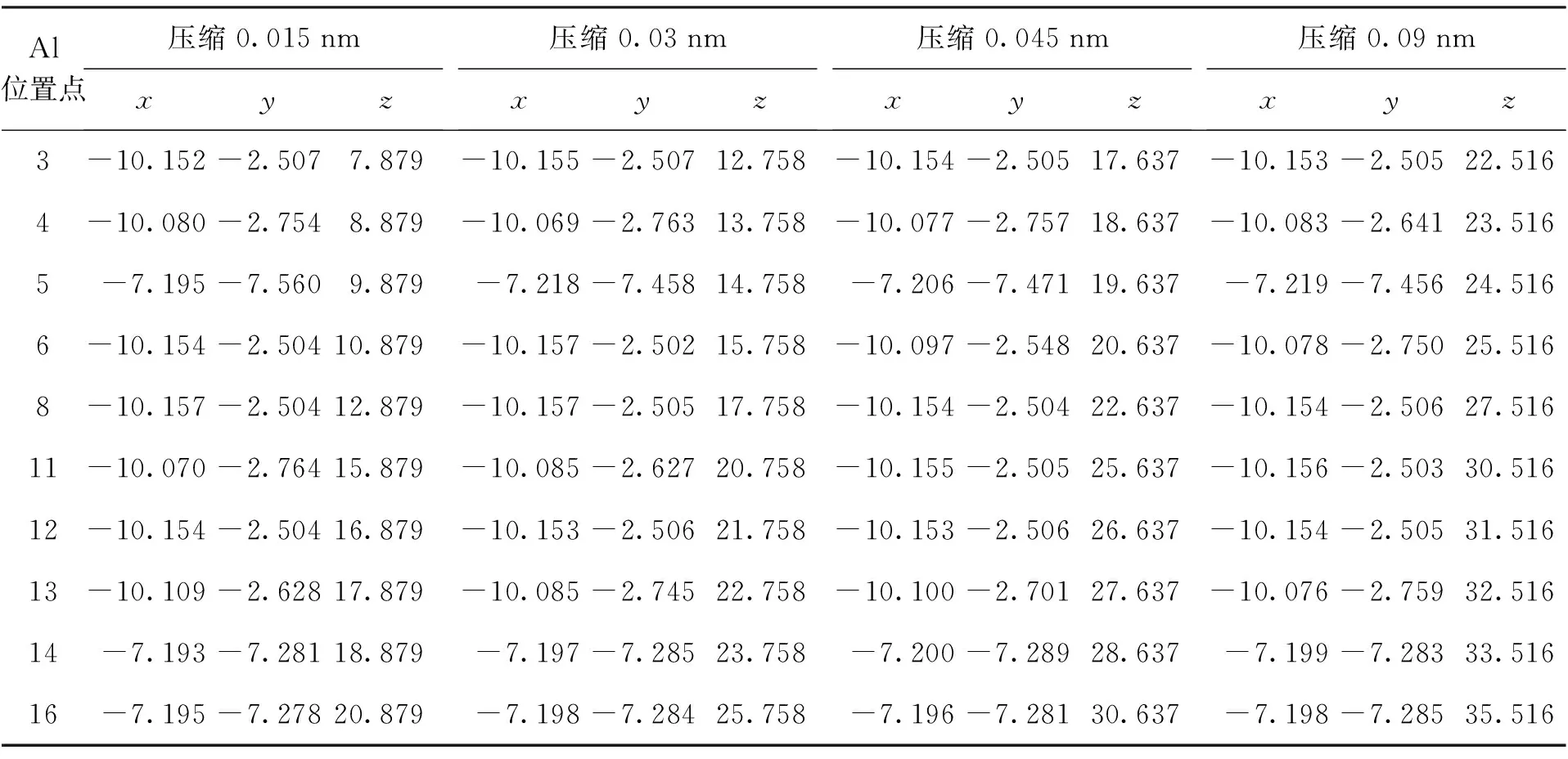
表4 不同压缩长度下Al的最终稳定吸附位置Tab.4 Final stable adsorption positions of Al under different compression lengths nm
B类初始Al吸附位置模型在压缩(5,5)CNT 0.015~0.09 nm的情况下,吸附能减小0.31~0.35 eV,吸附能减小约23%~25%,并且Al原子在CNT外壁最终稳定位置基本不变,与拉伸情况相似;压缩情况下,C类吸附位置除11点外吸附能减小0.25~0.26 eV,吸附能减小约21%~22%,11点处随着压缩量的增加,吸附能由减小0.26 eV提高到基本不变,稳定位置基本不变;A类吸附位置除6点外吸附能减小0.23~0.27 eV,吸附能减小约16%~18%,6点处随着压缩量的增加,吸附能由减小0.26 eV降低到减小0.53 eV.综上所述,对B(N)环状掺杂(5,5)CNT一定范围内的压缩会引起大部分初始Al吸附位置吸附能的降低.
3 结 论
基于密度泛函理论的第一性原理,研究了B(N)原子环状掺杂(5,5)CNT的掺杂效应,计算了其形成能,并对计算结果进行了分析.结果表明:B(N)环状掺杂(5,5)CNT的形成能是负值,说明掺杂是能量减少的过程,需要放出能量,所以反应可以进行,B(N)原子环状掺杂(5,5)CNT可以稳定存在.
研究了Al原子在掺杂B(N)(5,5)CNT表面吸附的结构及吸附能,结果表明,在离B(N)掺杂环最远的原子环上,是Al可以较稳定结合的位置.B(N)原子环状掺杂(5,5)CNT可以改善Al与CNT的结合.
在拉伸和压缩情况下的研究结果表明,一定范围内的拉伸会引起吸附能较强的Al原子在掺杂CNT表面吸附能的降低,并且稳定位置基本不变;而在吸附能较弱的Al原子在掺杂CNT表面吸附能有所提高,并且引起稳定位置向强吸附能位置转移.
[1]刘松,郭雪峰.基于单壁碳纳米管的功能分子电子器件研究 [J].化学学报,2013(4):478-484.
(LIU Song,GUO Xue-feng.Study on the functional molecular electronic devices based on single wall carbon nanotubes [J].Acta Chimica Sinica,2013(4):478-484.)
[2]张坚,李德英,赵龙志.碳纳米管/金属复合材料的研究现状与动向 [J].机械设计与制造,2011(2):102-106.
(ZHANG Jian,LI De-ying,ZHAO Long-zhi.Research status and trends of mental matrix composite reinforced by carbon nanotubes [J].Mechanical Design and Manufacturing,2011(2):102-106.)
[3]葛敏.碳纳米管及其金属复合材料拉伸行为的分子动力学模拟 [D].天津:天津大学,2011.
(GE Min.Dynamic’s simulations of molecules of carbon nanotubes and metal composites tensile beha-vior [D].Tianjin:Tianjin University,2011.)
[4]杨忠华.碳纳米管增强镁基复合材料力学性能有限元分析 [D].沈阳:沈阳工业大学,2011.
(YANG Zhong-hua.Finite element analysis of mechanical properties of carbon nanotube reinforced magnesium matrix composites [D].Shenyang:Shen-yang University of Technology,2011.)
[5]康建立.铜基体上原位合成碳纳米管(纤维)及其复合材料的性能 [D].天津:天津大学,2010.
(KANG Jian-li.Carbon nanotube (fiber) in situ synthesis on copper matrix of and properties of the composites [D].Tianjin:Tianjin University,2010.)
[6]朱文亮.界面相特性对碳纳米管增强复合材料力学性能的影响分析 [D].广州:华南理工大学,2011.
(ZHU Wen-liang.Analysis of the influence of inter phase properties on the mechanical properties of carbon nanotube reinforced composites [D].Guang-zhou:South China University of Technology,2011.)
[7]刘贵立,谢萌.碳纳米管增强钛基复合材料的界面应力分布 [J].沈阳工业大学学报,2015,37(6):684-689.
(LIU Gui-li,XIE Meng.Stress distribution at interface of carbon nanotubes reinforced titanium matrix composites [J].Journal of Shenyang University of Technology,2015,37(6):684-689.)
[8]胡雅婷,周硕,马晓兰,等.单壁碳纳米管储氢过程的GULP模拟 [J].人工晶体学报,2012(1):215-220.
(HU Ya-ting,ZHOU Shuo,MA Xiao-lan,et al.GULP simulation of hydrogen storage in single walled carbon nanotubes [J].Journal of Synthetic Crystals,2012(1):215-220.)
[9]沈超,胡雅婷,周硕,等.单壁碳纳米管低温及常温下储氢行为的模拟计算研究 [J].物理学报,2013,62(3):470-479.
(SHEN Chao,HU Ya-ting,ZHOU Shuo,et al.Simula-tion and calculation of hydrogen storage behavior of single wall carbon nanotubes at low temperature and room temperature [J].Acta Physica Sinica,2013,62(3):470-479.)
[10]闫瑞芳.高含量CNTs混杂增强2024Al基复合材料的制备及性能研究 [D].哈尔滨:哈尔滨工业大学,2012.
(YAN Rui-fang.Preparation and properties of high content CNTs hybrid reinforced 2024Al matrix composites [D].Harbin:Harbin Institute of Technology,2012.)
[11]邓春锋,马艳霞,薛旭斌,等.碳纳米管增强2024铝基复合材料的力学性能及断裂特性 [J].材料科学与工艺,2010,18(2):229-232.
(DENG Chun-feng,MA Yan-xia,XUE Xu-bin,et al.Mechanical properties and fracture properties of 2024 aluminum matrix composites reinforced by carbon nanotubes [J].Materials Science and Technology,2010,18(2):229-232.)
[12]徐慧,肖金,欧阳方平.扶手椅型单壁碳纳米管中的B/N对共掺杂 [J].物理学报,2010,59(6);4186-4193.
(XU Hui,XIAO Jin,OUYANG Fang-ping.Armchair single walled carbon nanotubes in the Co doped B/N [J].Acta Physica Sinica,2010,59(6);4186-4193.)
[13]刘贵立,姜艳,谢萌.碳纳米管超晶格结构的第一性原理 [J].沈阳工业大学学报,2015,37(3):289-293.
(LIU Gui-li,JIANG Yan,XIE Meng.First principle for carbon nanotube super lattice structure [J].Journal of Shenyang University of Technology,2015,37(3):289-293.)
[14]唐玉琴.Eu(Ⅲ)和Cu(Ⅱ)在多壁碳纳米管上的吸附 [D].兰州:兰州大学,2011.
(TANG Yu-qin.Adsorption of Eu (III) and Cu (II) on multi wall carbon nanotubes [D].Lanzhou:Lanzhou University,2011.)
[15]张变霞,杨春,冯玉芳,等.碳纳米管吸附铜原子的密度泛函理论研究 [J].物理学报,2009,58(6):4066-4071.
(ZHANG Bian-xia,YANG Chun,FENG Yu-fang,et al.Density functional theory study of copper atom adsorbed on carbon nanotubes [J].Acta Physica Sinica,2009,58(6):4066-4071.)
[16]张国英,张辉,魏丹,等.碳纳米管增强铝基复合材料电子理论研究 [J].物理学报,2007,56(3):1581-1584.
(ZHANG Guo-ying,ZHANG Hui,WEI Dan,et al.Study on the electronic theory of carbon nanotube reinforced aluminum matrix composites [J].Acta Physica Sinica,2007,56(3):1581-1584.)
[17]王昆鹏,师春生,赵乃勤,等.B(N)掺杂单壁碳纳米管的Al原子吸附性能的第一性原理研究 [J].物理学报,2008,57(12):7833-7840.
(WANG Kun-peng,SHI Chun-sheng,ZHAO Nai-qin,et al.First-principle study of the effect of boron (nitrogen)-doping on absorbing characteristics of aluminum on single-walled carbon nanotubes [J].Acta Physica Sinica,2008,57(12):7833-7840.)
(责任编辑:景勇英文审校:尹淑英)
Effect of tension and compression deformation on adsorption properties between Al and carbon nanotubes with B(N) doping
LIU Gui-li, SONG Yuan-yuan, JIANG Yan, ZHOU Shuang, WANG Tian-shuang
(School of Architecture and Civil Engineering, Shenyang University of Technology, Shenyang 110870, China)
In order to research the B(N) ring doping effect for (5, 5) carbon nanotubes (CNT) and the effect of tension and compression deformation on the adsorption properties between Al and CNT with B(N) ring doping, the geometry optimization for the Al adsorption model of CNT with B(N) ring doping was performed with both plane wave pseudo potential and generalized gradient approximation methods. In addition, the formation energy of CNT with B(N) ring doping was calculated, and the most stable adsorption positions of Al atoms were determined. The results reveal that the structure of (5, 5) CNT with B(N) ring doping is stable. Moreover, the B(N) ring doping can increase the adsorption energy between CNT and Al. However, the tensile and compression deformation within a certain range will reduce most adsorption energy between CNT and Al.
carbon nanotube(CNT); first-principle; doping; Al atom adsorption; tensile and compression deformation; adsorption energy; density functional theory; geometry optimization
2015-11-02.
国家自然科学基金资助项目(51371049).
刘贵立(1963-),男,山东济宁人,教授,博士,主要从事建筑结构与材料可靠性、机械结构损伤与诊断等方面的研究.
10.7688/j.issn.1000-1646.2016.04.06
TB 383
A
1000-1646(2016)04-0391-06
*本文已于2016-05-12 13∶56在中国知网优先数字出版. 网络出版地址: http:∥www.cnki.net/kcms/detail/21.1189.T.20160512.1356.010.html
