水产品中甲基汞测定方法的优化
2016-09-13柯常亮陈洁文林颂雄纪晓敏李刘冬
刘 奇,柯常亮,陈洁文,林颂雄,蔡 楠,黄 珂,纪晓敏,李刘冬,*
(1.中国水产科学研究院南海水产研究所,农业部水产品加工重点实验室,农业部水产品贮藏保鲜质量安全风险评估实验室(广州),广东广州 510300;2.汕尾市海洋与渔业环境监测站,广东汕尾 516611)
水产品中甲基汞测定方法的优化
刘奇1,柯常亮1,陈洁文1,林颂雄1,蔡楠1,黄珂1,纪晓敏2,李刘冬1,*
(1.中国水产科学研究院南海水产研究所,农业部水产品加工重点实验室,农业部水产品贮藏保鲜质量安全风险评估实验室(广州),广东广州 510300;2.汕尾市海洋与渔业环境监测站,广东汕尾 516611)
本研究对标准方法进行了优化,建立了水产品中甲基汞残留的快速测定方法。本方法采用硫酸和溴化钾超声辅助提取样品中甲基汞,经甲苯反萃,气相色谱测定。结果显示,在25.0~1000 μg/L浓度范围内,甲基汞标准曲线方程为y=21.49x-660.9,线性良好(r=0.9995);方法检测限和定量限分别为7.58、25.0 μg/kg;加标量为50.0、100、500、1000 μg/kg时,回收率范围为80.8%~108%,相对标准偏差范围为1.56%~8.10%,表明该方法满足水产品中甲基汞残留测定的要求。方法优化后步骤减半,耗时缩短三分之二,样品分析效率大幅提高,适合批量测定水产品中甲基汞残留。
甲基汞,水产品,气相色谱法,快速测定
甲基汞是一种剧毒有机重金属污染物,可对人体造成以神经系统为主的全身性伤害[1],并有致癌作用[2]。甲基汞污染引起的日本水俣病事件是世界八大公害事件之一[3]。水体和底泥中70%以上的汞以甲基汞的形式存在[4],由于分子量小、脂溶性强,甲基汞极易在水产品中富集,经食物链进入人体[5]。研究表明,水产品是人类甲基汞暴露的主要来源[6]。为保障人体健康,联合国粮农组织(FAO)和世界卫生组织(WHO)规定,水产品中肉食鱼类和非肉食鱼类甲基汞限量分别为1.0、0.5 mg/kg,中国、美国等采纳了该标准[7-8];2015年起,欧盟将水产品及水生生物中汞和汞类化合物环境质量标准(EQS)设为20 μg/kg[9],进一步加强对甲基汞的危害控制。近年来,我国加强了食品质量安全的监管,水产品中甲基汞残留的监控覆盖面和样品数量大幅增加,对分析方法的要求也越来越高。
水产品中甲基汞残留测定方法是近年来研究热点。气相色谱-质谱联用法(GC-MS)[10]、原子荧光法(AFS)[11-12]、电感耦合等离子体质谱法(ICPMS)[13-14]和气相色谱法[15]等现代仪器分析技术被广泛应用于甲基汞检测研究。其中,GC-MS法灵敏度高,但需要四乙基硼化钠(NaBEt4)等试剂进行衍生,操作复杂,且NaBEt4室温下遇到含有机物的蒸汽就会燃烧爆炸,危险性高。AFS及ICPMS在定性和定量方面都具有较好的准确度和灵敏度,但需与色谱串联,仪器复杂且价格昂贵。我国标准中水产品甲基汞残留测定方法为气相色谱法,该法具有灵敏度高、仪器价格低廉等优势,在实际工作中被广泛应用;但其前处理操作繁杂,对色谱柱及巯基棉柱要求高,结果不稳定[16],大批量样品测定较难操作,需进一步优化改进。
本研究对甲基汞残留检测气相色谱法样品前处理过程进行改进,建立了水产品中甲基汞残留量检测新方法,方法简便、准确、快速、环保且成本低,为水产品中甲基汞风险监测提供可靠的技术支持。
1 材料与方法
1.1仪器与试剂
气相色谱仪(配μECD检测器)7890A型,Agilent公司;色谱柱DB-5型(30 m×0.25 mm×0.25 μm),Agilent公司;电子天平XP205DR型,瑞士梅特勒公司;冷冻离心机DL-6000B型,上海安亭科学仪器厂。
氯化甲基汞标准物质(CAS 115-09-3)96.0%,Dr.Ehrenstorfer GmbH公司;甲苯色谱纯,Sigma-Aldrich公司;五水合硫酸铜(CuSO4·5H2O)、溴化钾、浓硫酸均为分析纯。
硫酸铜溶液(1.0 mol/L):称取62.5 g CuSO4·5H2O加纯水溶解定容至250 mL;溴化钾-硫酸混合溶液:称取180 g溴化钾溶于200 mL纯水中,取55.0 mL浓硫酸溶于100 mL纯水中,将两份溶液混合定容至500 mL。
1.2仪器条件
参考朱霞萍等[17]方法。进样口温度250 ℃;进样量1 μL,不分流进样;恒流模式,柱流速2 mL/min;μECD检测器温度300 ℃,尾吹气50 mL/min;初始柱温60 ℃,保持1 min,以20 ℃/min升至130 ℃,保持1 min,以30 ℃/min升至280 ℃,保持1 min。
1.3标准溶液配制
1.3.1标准储备溶液准确称取氯化甲基汞标准物质0.0626 g,以甲苯溶解定容至50.00 mL容量瓶,配成质量浓度为1000 mg/L的甲基汞标准储备溶液,2~5 ℃条件下可储存1年。
三、总之,玉米粗缩病是具有毁灭性的病害,只要发生就会很难治理,虽然在病株上喷施药剂但也难恢复正常生长。因此,加强在玉米粗缩病常发地区定点监测和预报,定期调查小麦、田间杂草和玉米粗缩病的病株率,同时也调查灰飞虱越冬基数、发生密度和带毒率,及时清除田间杂草,改变种植模式使用种子包衣,在科学的引导下农民集中使用药剂防治,将农业防治和化学防治相结合,才能有效控制灰飞虱和玉米粗缩病的发生,控制粗缩病的危害蔓延,尽可能的降低产量损失,提高玉米产量,增加农民收入。
1.3.2标准中间溶液准确移取甲基汞标准储备溶液1.00 mL,以甲苯定容至100.0 mL容量瓶,配成质量浓度为10.0 mg/L的甲基汞标准中间溶液,2~5 ℃条件下可储存6个月。
1.3.3标准使用溶液使用时,移取一定体积的甲基汞标准中间溶液,用甲苯配制成质量浓度分别为25.0、50.0、100、200、500、1000 μg/L的标准工作溶液。
1.4样品前处理
新鲜鱼、虾、贝取可食肌肉组织捣碎、混匀,称取2.00 g样品于50 mL具塞离心管中,加入1.00 mol/L硫酸铜溶液0.5 mL、纯水4.0 mL,涡旋混匀;向离心管中加溴化钾-硫酸混合溶液2.0 mL,涡旋混匀;250 W超声提取10 min;5500 r/min,4 ℃离心10 min,将上清液转移至另一50 mL具塞离心管。再向样品残渣加入1.00 mol/L硫酸铜溶液0.5 mL、纯水4.0 mL、溴化钾-硫酸混合溶液2.0 mL,按以上步骤重复提取一次,合并两次提取液。向提取液中加入4.0 mL甲苯,涡旋混合,静置分层,吸取1.0 mL上层甲苯于进样瓶中,用于气相色谱仪分析。
2 结果与讨论
2.1样品前处理
甲基汞在生物组织中易与硫结合,必须破坏硫化合物并将其转化为卤代甲基汞,才可将甲基汞提取至有机溶剂中[18]。Hintelmann[19]研究表明,硫酸铜、硫酸和溴化钾混合溶液对甲基汞的提取效果优于盐酸、氢氧化钾等提取液,且产生乙基汞等杂质较少。本研究利用Cu2+置换出组织中的甲基汞,通过溴化钾-硫酸混合溶液将甲基汞转化为溴化甲基汞,以达到对样品中的甲基汞提取分离的目的。图1为在本实验条件下得到的浓度为50.0 μg/L甲基汞标准溶液(A)和加标量为100 μg/kg的牡蛎样品(B)气相色谱图。从图中可以看出,本方法对样品处理后得到的甲基汞色谱峰峰型尖锐、对称,信号响应高,基体干扰小。硫酸铜、硫酸和溴化钾混合溶液对甲基汞的提取和纯化达到了满意效果。
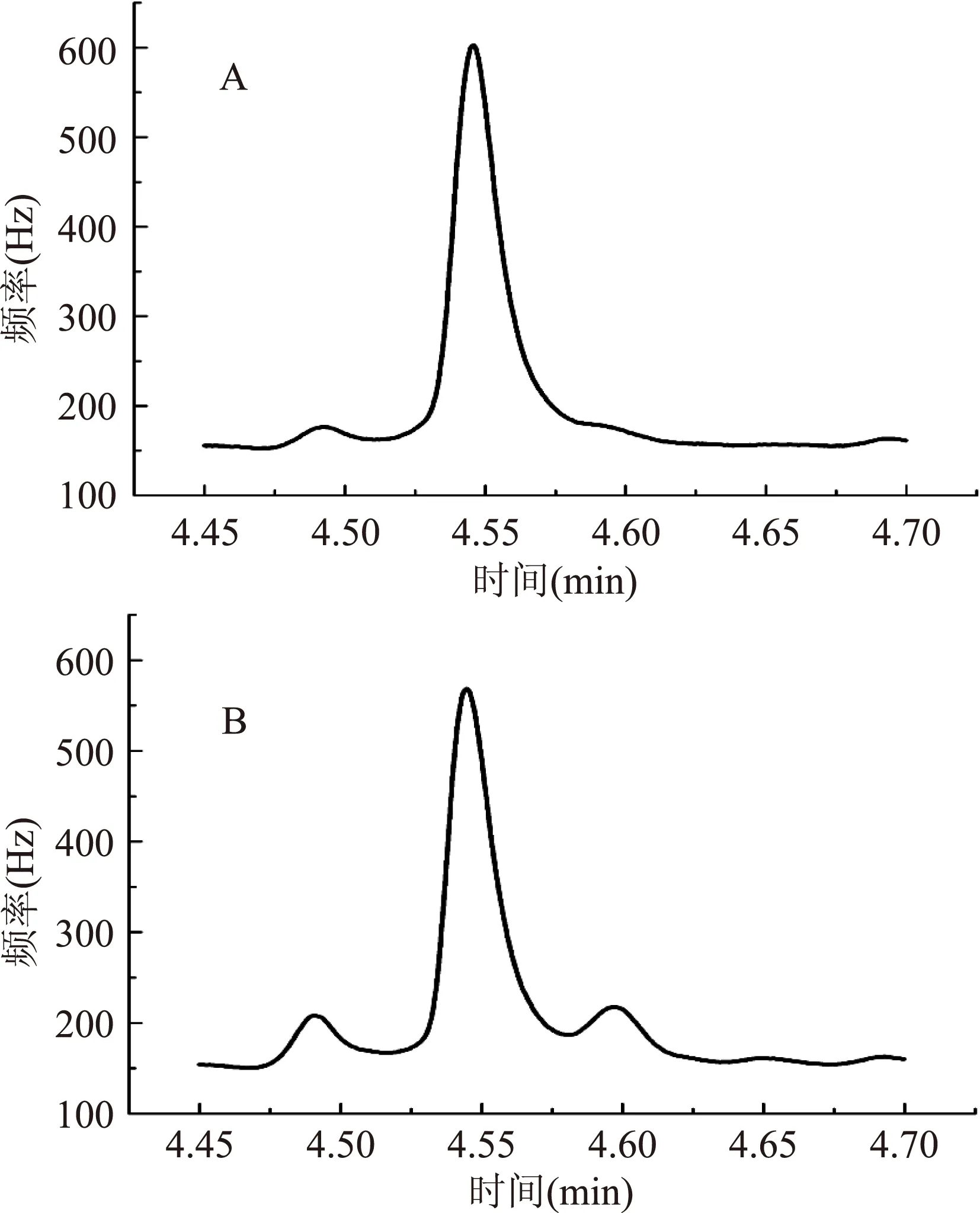
图1 甲基汞标准品(A)和牡蛎样品(B)气相色谱图Fig.1 Gas chromatograms of MMC standard solution(A) and 100 Oyster tissue spiked with MMC(B)

表1 GB/T5009.17-2003和本方法甲基汞检测前处理过程比较Table 1 Comparison of the preparation procedure for MMC determination using GB/T5009.17-2003 or primary method
注:表中所列耗时为作者处理一个实验样品所需时间,结果因操作不同可能存在差异。
本研究用毒性较小的甲苯代替苯作为萃取剂[20],降低了实验对环境和操作人员健康造成的危害。同时,本研究在反萃过程中采用5500 r/min高速离心处理,避免乳化现象,有助于提高回收率。
此外,本研究采用超声10 min辅助提取样品中的甲基汞,各样品回收率可达80%以上(表2);与振荡提取60 min处理[16]相比,本方法不仅能提高提取速率,也能增大提取效率。
2.2方法效率评估
GB/T 5009.17-2003处理一个样品至少需12步,经3次转移和1次过柱与洗脱,共耗时约100 min(表1),其操作过程繁杂,尤其巯基棉富集过程容易导致结果重现性欠佳[16]。本研究前处理过程只需7步,经1次转移过程,单个样品耗时仅约30 min,前处理简便、快速。本方法所有操作在离心管中完成,避免了多次转移对目标物造成的损失,保证了回收率,也节约了实验试剂和材料,降低成本。
此外,GB/T5009.17-2003法需在实验前制备巯基棉,整个制备过程约需5 d,制备过程对温度、水分要求较高,且巯基棉吸附效率对实验结果影响较大。本研究实验前只需简单的溶液配制,操作简单。
2.3线性关系
将标准工作溶液进气相色谱测定,以峰面积为纵坐标y,以甲基汞的质量浓度为横坐标x,作线性回归分析并绘制标准曲线(图2)。所得线性方程为y=21.49x-660.9,线性相关系数r=0.9995,甲基汞在25.0~1000 μg/L浓度范围内有良好线性。

图2 甲基汞标准工作曲线Fig.2 Calibration curve for MMC determination
2.4检出限与定量限
本研究以甲基汞添加量为50.0 μg/kg的空白样品考察方法灵敏度。加标样品按上述方法进行提取、净化、测定并计算信噪比。以3倍信噪比(S/N=3)时对应的添加浓度为检出限,以10倍信噪比(S/N=10)对应的添加浓度为定量下限,依此计算得出,本方法检出限为7.58 μg/kg,定量限为25.0 μg/kg,陈雪昌等[1]利用高效液相色谱-原子荧光法测定水产品中甲基汞定量限为20 μg/kg,灵敏度同本法相近。该法检出限远低于国内标准规定限量[7],结果灵敏可靠,可以满足甲基汞检测工作等需求。
2.5回收率与精密度
本研究以草鱼、卵形鲳鲹、对虾、牡蛎等4种代表性水产品进行加标实验(表2),空白加标样品中未检出甲基汞。每种水产品分别做4个浓度水平,每个水平做6个重复,按上述方法测定样品中的甲基汞含量,考察方法的回收率和精密度。结果显示(表2),4种水产品在加标浓度为50.0、100、500、1000 μg/kg时,回收率范围为80.8%~108%,相对标准偏差范围为1.56%~8.10%。由此可见,本方法测定4种不同水产品基质中甲基汞都有较高的回收率和良好的精密度,可以满足水产品中甲基汞残留检测要求。

表2 不同水产品加标回收率与精密度(n=6)Table 2 Recovery and precision for MMC determination in different aquatic product(n=6)
3 结论
本研究对甲基汞气相色谱法前处理过程进行优化,改进后的方法甲基汞提取和净化过程简便、快速、环保、节约,检测结果杂质干扰少,在25.0~1000 μg/L浓度范围内线性良好,方法灵敏、可靠,回收率高,精密度和准确度都可以满足实际工作中大量样品检测需求。
[1]陈雪昌,梅光明,张小军,等. 高效液相色谱-原子荧光法测定水产品中甲基汞含量. 食品科学,2012,33(4):234-237.
[2]蔡文洁,江研因. 甲基汞暴露健康风险评价的研究进展[J]. 环境与健康杂志,2008,25(1):77-81.
[3]郑伟刚,黎中宝,李文静,等. Hg2+对5种鳗鲡白仔急性毒性影响的研究[J]. 南方水产科学,2011,7(4):177-188.
[4]尚晓虹,赵云峰,张磊,等. 水产品中甲基汞测定的液相色谱-原子荧光光谱联用方法的改进[J]. 色谱,2011,29(7):667-672.
[5]李新贵,窦强,黄美荣. 共聚苯胺吸附剂对含汞废水的处理[J]. 南方水产,2007,3(6):8-13.
[6]Li P,Feng X B,Yuan X B,et al. Rice consumption contributes to low level methylmercury exposure in southern China[J]. Environment International,2012,49(1):18-23.
[7]中华人民共和国卫生部. GB 2762-2012食品安全国家标准:食品中污染物限量[S]. 北京:中国标准出版社,2012.
[8]Ariane V Z,Sergio C,Carmen I P,et al. Method development for the simultaneous determination of methylmercury and inorganic mercury in seafood[J]. Food Control,2014,46(1):351-359.
[9]Miniero R,Becalonie E,Cafere M,et al. Mercury(Hg)and methyl mercury(MeHg)concentrations in fish from the coastal lagoon of Orbetello,central Italy[J]. Marine Pollution Bulletin,2013,76(1):365-369.
[10]Beceiro E,Guimaraes A,Alpendurada M F. Optimisation of a headspace-solid-phase micro-extraction method for simultaneous determination of organometallic compounds of mercury,lead and tin in water by gas chromatography-tandem mass spectrometry[J].Journal of Chromatography A,2009,1216(29):5563-5569.
[11]Guo Y N,Feng X B,Li Z G,et al. Distribution and wet deposition fluxes of total and methyl mercury in Wujiang River
Basin,Guizhou,China[J]. Atmospheric Environment,2008,42(1):7096-7103.
[12]肖亚兵,崔颖,陈文硕,等.液相色谱-原子荧光法测定鱼肉中有机汞形态[J]. 食品研究与开发,2014,35(21):106-108.
[13]Jagtap R,Krikowa F,Maher W,et al. Measurement of methyl mercury(I)and mercury(II)in fish tissues and sediments by HPLC-ICPMS and HPLC-HGAAS[J].Talanta,2011,85(1):49-55.
[14]Heidi P,Paavo P,Juha P,et al. Determination of low methylmercury concentration in peat soil samples by isotope dilution GC-ICP-MS using distillation and solvent extraction[J].Chemosphere,2015,124(1):47-53.
[15]中华人民共和国卫生部. GB/T 5009.17-2003食品中总汞及有机汞的测定[S]. 北京:中国标准出版社,2003.
[16]刘素琴,牟晓崟. 气相色谱法测定水产品中痕量甲基汞的方法研究[J]. 中国卫生检验杂志,2015,25(3):344-347.
[17]朱霞萍,张勇,汪模辉,等. 毛细管气相色谱法测定生物样品中痕量甲基汞[J]. 理化检验,2006,42(5):344-346.
[18]王嵩,李宏林,胡明明,等. 甲基汞检测研究进展[J]. 化学研究与应用,2011,23(2):129-136.
[19]Hintelmann H. Comparison of different extraction techniques used for methylmercury analysis with respect to accidental formation of methylmercury during sample preparation[J]. Chemosphere,1999,39(7):1093-1105.
[20]宗万里. 气相色谱-质谱联用法检测烤鱼片中甲基汞含量[J]. 生命科学仪器,2014,12(1):45-47.
Method optimization for methylmercury determination in aquatic products
LIU Qi1,KE Chang-liang1,CHEN Jie-wen1,LIN Song-xiong1,CAI Nan1,HUANG Ke1,JI Xiao-min2,LI Liu-dong1,*
(1.South China Sea Fisheries Research Institute,Chinese Academy of Fishery Sciences,Key Laboratory of Aquatic Product Processing,Ministry of Agriculture,Laboratory of Quality and Safety Risk Assessment for Aquatic Product on Storage and Preservation,Ministry of Agriculture,Guangzhou 510300,China;2. Shanwei Oceanic and Fishery Environment Monitoring Station,Shanwei 516611,China)
A method was developed for methylmercury(MMC)fast determination in aquatic products through standard method optimizing. Samples were extracted by sulfuric acid and potassium bromide in assist of ultrasound,back-extracted by toluene,detected by gas chromatography(GC). As shown,calibration curve(y=21.49x-660.9)was in good linearity with a correlation coefficient of 0.9995 in the range of 25.0~1000 μg/L. The limit of detection(LOD)and the limit of quantitation(LOQ)were 7.58 and 25.0 μg/kg,respectively. While spiked at 50.0,100,500 and 1000 μg/kg,the recoveries of MMC ranged from 80.8% to 108% with relative standard deviation(RSD)of 1.56%~8.10%. It indicated that the method was qualified to detect MMC residue in aquatic products. After optimization,efficiency of sample analysis was greatly improved because of the reduction of half procedures and two thirds time saving,which made the method suitable for quantitation of MMC in batch samples.
methylmercury;aquatic products;GC;fast determination
2015-04-13
刘奇(1987-),男,硕士研究生,研究实习员,研究方向:水产品质量安全,E-mail:liuqi03@hotmail.com。
李刘冬(1959-),男,本科,研究员,研究方向:水产品质量安全,E-mail:168LLd@163.com。
国家农产品质量安全风险评估重大专项(GJFP2014009);中央级公益性科研院所基本科研业务费专项(中国水产科学研究院南海水产研究所,2014TS10);农业部水产品加工重点实验室开放基金项目(NYJG201306)。
TS254.7
A
1002-0306(2016)03-0299-04
10.13386/j.issn1002-0306.2016.03.054
