HS+HO2气相反应机理及主通道速率常数的理论研究
2016-09-13张田雷杨晨凤旭凯王竹青王睿刘秋丽张鹏王文亮
张田雷 杨晨 凤旭凯 王竹青 王睿 刘秋丽 张鹏 王文亮
(1陕西理工学院化学与环境科学学院,陕西省催化基础与应用重点实验室,陕西汉中723001;2山东省科学院海洋仪器仪表研究所,山东省海洋环境监测技术重点实验室,山东青岛266001;3陕西师范大学化学化工学院,陕西省大分子科学重点实验室,西安710062)
HS+HO2气相反应机理及主通道速率常数的理论研究
张田雷1,*杨晨1凤旭凯1王竹青2王睿1刘秋丽1张鹏1王文亮3,*
(1陕西理工学院化学与环境科学学院,陕西省催化基础与应用重点实验室,陕西汉中723001;2山东省科学院海洋仪器仪表研究所,山东省海洋环境监测技术重点实验室,山东青岛266001;3陕西师范大学化学化工学院,陕西省大分子科学重点实验室,西安710062)
采用CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)双水平计算方法构建了HO2+HS反应体系的单、三重态反应势能面,并对该反应主通道的速率常数进行了研究。研究结果表明,标题反应经历了八条反应通道,其中三重态反应通道R 1是标题反应主通道。此通道包含路径Path 1(R→3IM1→3TS1→P1 (3O2+H2S))和Path 1a(R→3IM1a→3TS1a→P1(3O2+H2S))两条路径。利用经典过渡态理论(TST)与变分过渡态理论(CVT)并结合小曲率隧道效应模型(SCT),分别计算了主路径Path 1和Path 1a在200-800K温度范围内的速率常数kTST、kCVT和,在此温度区间内路径Path 1和Path 1a具有负温度系数效应。速率常数计算结果显示,对主路径Path 1和Path 1a而言,变分效应在计算温度段内有一定影响,与此同时量子力学隧道效应在低温段有显著影响。路径Path 1和Path 1a的CVT/SCT速率常数的三参数表达式分别为(200-800K)=1.54×10-5T-2.70exp(1154/T)cm3∙mo lecule-1∙s-1和200-800K)=5.82× 10-8T-1.84exp(1388/T)cm3∙mo lecule-1∙s-1。
HS;HO2;势能面;反应机理;速率常数
1 引言
富硫煤、含硫有机物燃烧以及含硫有机化合物的生物分解所产生的含硫有机小分子主要包括硫醚(如CH3SCH3(DMS))、硫醇(如CH3SH、MeSH)、二甲亚砜硫(CH3SOCH3(DMSO))等稳定物种以及HS、CH3S、CH3SO、CH3SS、CH3SOO、CH3SCH2、CH3SCH2O、CH3CH2S、CH3SSCH3等不稳定物种1。该类含S化合物虽然不是地球大气环境的主要组成部分,但对气候变化、酸雨形成、大气硫循环等一系列环境问题有着显著的影响2-5。作为含硫小分子化合物的重要组成之一,HS自由基不仅是煤炭中硫转化过程所产生的关键中间体,也是大气光化学反应的主要产物之一,在大气环境污染中扮演着重要的角色6。因此,有关HS与大气中其它物种的反应,在实验和理论方面已有不少文献报道,例如HS与Cl7、NO8、NO29、N2O10、O39、O211、Cl212、CO13、CH314、(CH3)2O15、CHF316和C2H217的物种的反应等。通过已有研究得到的含硫有机小分子大气化学反应机理及动力学性质,能极大提高大气化学反应动力学研究水平,推动认识大气S循环、消除S化合物的大气污染。但遗憾的是,有关HS自由基与HO2自由基的反应目前在实验和理论方面均未见报道。
事实上,过氧自由基(HO2)是继OH自由基之后大气环境中最重要的自由基,研究该自由基的形成与消耗机制一直以来都是化学和环境方面的研究热点18-20。基于此,研究HS自由基与HO2自由基的大气反应机理及其动力学性质,对解决环境污染和保护大气环境均具有重要的指导意义。本文采用CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+ G(2df,2p)双水平计算方法,构造了如示意图1所示的HO2+HS单、三重态反应势能剖面图,讨论了抽氢及抽氧机理,并计算了反应主通道在200-800K温度范围内的速率常数。
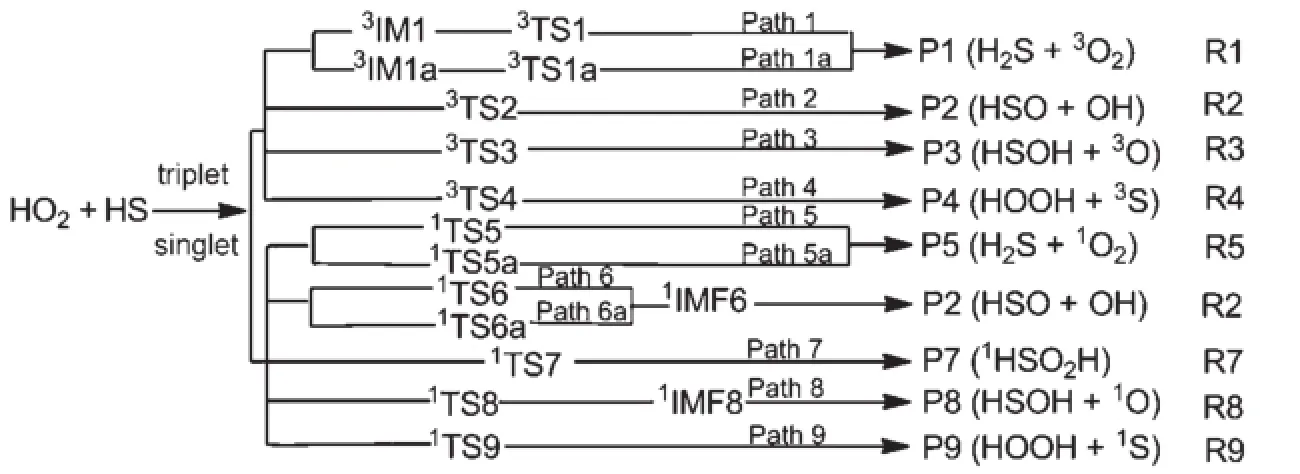
示意图1 HO2+HS反应通道示意图Scheme1 Schematic diagram for the HO2+HS reaction channels
2 计算方法
在B3LYP/6-311+G(2df,2p)水平上对HO2+HS反应各通道中所包含的反应物、中间体、过渡态以及产物进行了全参数几何构型优化,确认了各稳定点和过渡态的几何构型,并在相同水平上进行了振动频率分析,确定了优化得到的几何构型是对应于势能面上的极小点和过渡态,并得到零点能和热力学参数。此外在上述相同水平上进行了内禀反应坐标(IRC)21分析,验证了各过渡态与对应反应物(前中间体)和产物的关联性。为了获得更加精确的最小能量路径(VMEP(s)),采用双水平计算方法CCSD(T)22/6-311++G(3df,2pd)//B3LYP/6-311+ G(2df,2p)对所有驻点物种以及各IRC路径上的选择点进行了单点能校正。由于B3LYP方法的可靠性已在HO2与CH2S和H2S反应中得到证实,进一步由于标题反应的原理与其类似,因此在本文中采用了该方法23,24。上述计算均在Gaussian 09程序25完成。
应用VKLab程序包26,分别采用经典过渡态理论(TST),与变分过渡态理论(CVT)27并结合小曲率隧道效应模型(SCT)28对HO2+HS反应主通道的速率常数进行了计算。标题反应主(如(1)式所示)通道,与甲醛和过氧自由基气相反应机理类似29,首先形成一个无能垒的中间体(IM),然后分别经过渡态(TS)越过一个适中的能垒生成产物(P)。

在(1)式中假设中间体IM与反应物(HO2+HS)处于平衡状态,根据稳态条件,(1)式的速率常数k如(2)式所示。

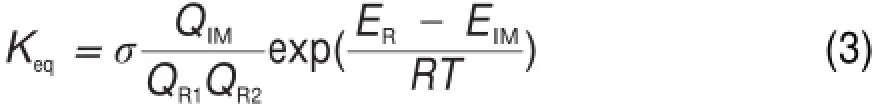
(2)式中反应第一步的平衡常数Keq可由(3)式计算而来,反应第二步的速率常数k2可通过VKLab程序26计算得出。(3)式中反应物配分函数QR1和QR2以及中间体配分函数QIM由VKLab程序26计算所得;ER和EIM是CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上反应物、中间体的总能量;σ是对称数。这种计算速率常数的方法的有效性在其它文章也有报道30。
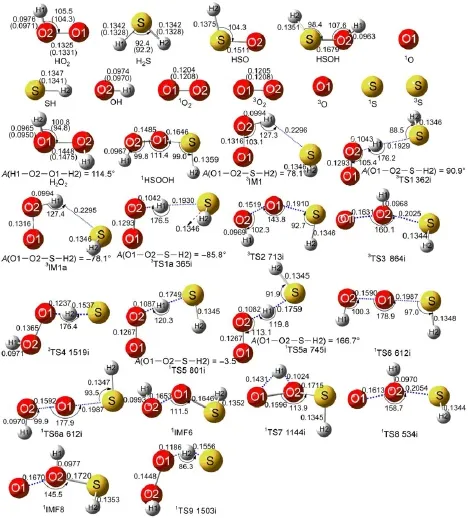
图1 B3LYP/6-311+G(2df,2p)水平上优化所得的各中间体、过渡态和产物的几何构型及部分实验值aFig.1 Optim ized geometricalstructures for all the stationary pointsand transition statesat the B3LYP/6-311+G(2df,2p) levelalong w ith the availab le experim ental valuesabond length in nm,bond angle in degree;A:dihedralangle.aThe values in parenthesesare theexperimentalvaluesand taken from Ref.31.
3 结果与讨论
在B3LYP/6-311+G(2df,2p)水平上优化的反应物、中间体、过渡态和产物以及可获得物种的实验数据31见图1。本文所涉及的单、双和三重态物种的〈S2〉(自旋角动量平方算符本征值)分别集中在0.001-0.002、0.752-0.756和2.001-2.003之间,消除自旋污染后的〈S2〉值与本征值0.000、0.750和2.000完全吻合,说明B3LYP/6-311+G(2df,2p)水平上所得的波函数污染并不严重。通过与图1所示的实验值比较,发现在B3LYP/6-311+G(2df,2p)水平上的计算值与相应的实验值吻合的很好,键长和键角的平均偏差分别为0.0007 nm和0.7°,说明本文选择B3LYP/6-311+G(2df,2p)方法优化几何构型是合适的。表1列出了CCSD(T)/6-311++G(3df, 2pd)//B3LYP/6-311+G(2df,2p)水平上HO2+HS反应各驻点物种的相对能、焓和吉布斯自由能等信息,其中生成产物3O2+H2S的反应焓为-174.91 kJ∙mol-1,与实验值((-173.32±3.25)kJ∙mol-1)31比较接近。为了进一步证明CCSD(T)/6-311++G(3df, 2pd)方法计算标题反应单点能的可靠性,表1给出了产物通道(3O2+H2S)在CCSD(T)/aug-cc-pVTZ// B3LYP/6-311+G(2df,2p)水平上所有物种相对能(∆E)、焓变(ΔH)和吉布斯自由能变(ΔG)。从表1中可以看出在CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上HO2+HS反应中各中间体、过渡态和产物相对于反应物的能量比CCSD(T)/ aug-cc-pVTZ//B3LYP/6-311+G(2df,2p)水平上相对应的值改变了0.70-2.33 kJ∙mol-1,进一步证实本文选用CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+ G(2df,2p)方法研究HO2+HS气相反应机理是合理的。因此本文将采用CCSD(T)/6-311++G(3df,2pd)// B3LYP/6-311+G(2df,2p)双水平计算方法对图2所绘的标题反应势能面进行理论研究。
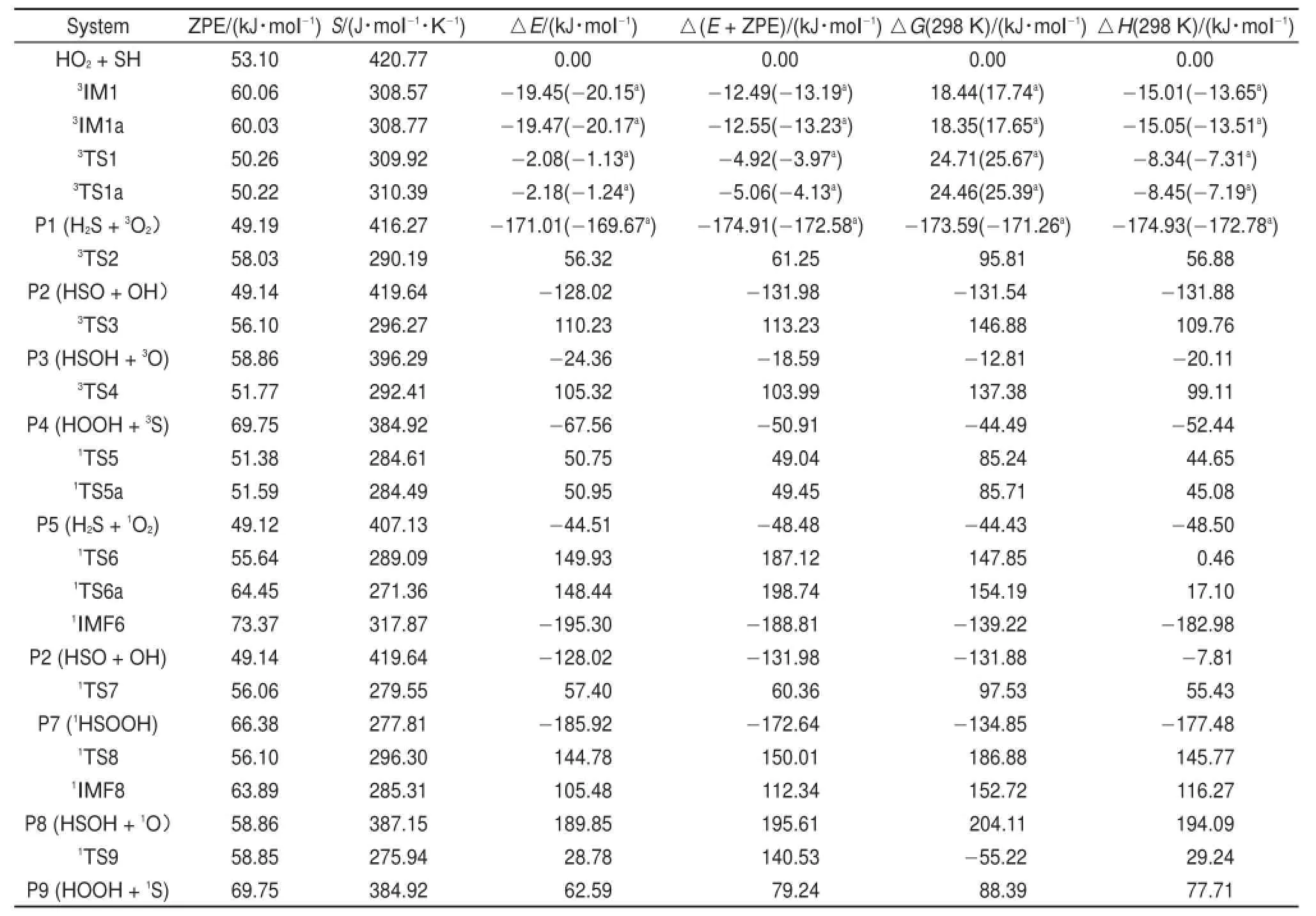
表1 在CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上HO2+HS反应各驻点物种的零点能(ZPE)、熵(S)、相对能(ΔE和Δ(E+ZPE))、吉布斯自由能变(ΔG(298K))和焓变(ΔH(298K))Tab le 1 Zero-poin t energy(ZPE),en trop ies(S),relative energies(ΔE andΔ(E+ZPE)),free energies(ΔG(298 K))and enthalpies(ΔH(298K))for the HO2+HS reactionat the CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)levelof theory
3.1HO2+HS反应机理
B3LYP/6-311+G(2df,2p)计算结果表明,HS中S的自旋密度为0.97,说明不成对电子主要集中在S上,HO2中端基氧原子O1与非端基氧原子O2的自旋密度分别为0.74和0.25,即不成对电子主要定域在两个氧原子上。考虑到HO2中H原子的高活性,可以推测,HO2+HS的反应过程中,主要存在如下作用方式:(1)HS中的S抽取HO2中的H;(2)HS中的S抽取HO2端基氧原子O1或非端基氧原子O2;(3)HO2端基氧原子O1抽取HS中的H。与HO2+OH反应32-34相比较,HS中的S抽取HO2中的OH部分以及HO2端基氧原子抽取HS中的H是两种新的进攻方式,在HO2+OH反应体系的先前研究中未见报道。本文以下部分将对HO2+HS反应过程中存在的上述作用方式所产生的8条反应通道进行研究。
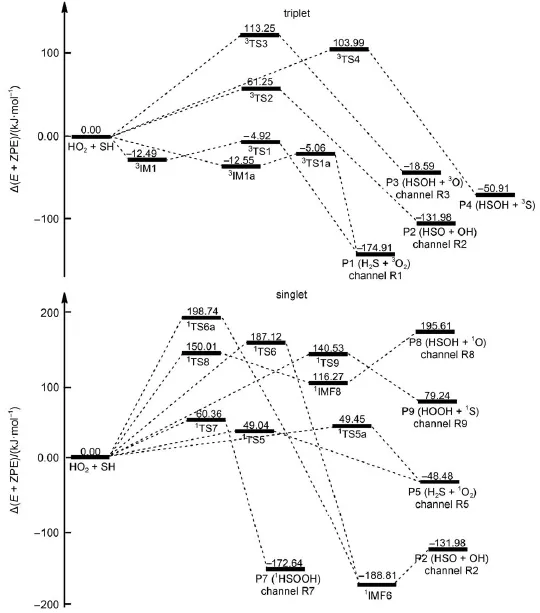
图2 CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)+ZPE水平上HO2+HS反应势能剖面图Fig.2 Schem atic energy d iagram s in HO2+HS reaction at the CCSD(T)/6-311++G(3df,2pd)// B3LYP/6-311+G(2df,2p)+ZPE level
3.1.1三重态HS+HO2反应势能面
三重态HS+HO2反应势能面上产生了4条反应通道R1、R2、R3和R4(见示意图1与图2),分别得到了产物P1(H2S+3O2)、P2(HSO+OH)、P3 (HSOH+3O)和P4(HOOH+3S)。其中通道R3可以看成是HS中的S与HO2O1中HO2部分的耦合形成过渡态3TS3,此过程需克服高达113.23 kJ∙mol-1的势能。此外通道R4可以看成是HO2端基氧原子O1抽取HS中的H经过渡态3TS4生成P4(HOOH+3S),该过程需克服103.99 kJ∙mol-1的势能。与通道R1和R2相比,通道R3和R4在三重态反应通道中势垒较高。因此,从动力学角度而言,生成P3和P4的几率是4条三重态反应通道R1、R2、R3和R4中较小的。因此本文仅对三重态反应通道R1和R2进行分析讨论。
通道R1是由HS中的S原子抽取HO2中的H原子所得,该通道包括路径Path 1和Path 1a两条路径。对于路径Path 1和Path 1a而言,首先分别形成三重态中间体3IM1和3IM1a。3IM1和3IM1a的稳定化能分别为12.49和12.55kJ∙mol-1,它们的稳定性可能与其结构中均存在S…H1氢键有关。中间体3IM1和3IM1a的差别在于O1―O2与S―H2排列取向不同,3IM1中O1―O2―S―H2对应的二面角为78.1°。3IM1a中O1―O2―S―H2对应的二面角为-78.1°。从中间体3IM1和3IM1a出发,分别经过渡态3TS1和3TS1a形成产物P1(H2S+3O2)。由图2可知,过渡态3TS1和3TS1a的相对能量分别为-4.92和-5.06kJ∙mol-1。此外,生成P1的反应吉布斯自由能为-174.91 kJ∙mol-1,表明三重态反应通道R1所包含的两条路径Path 1和Path 1a均较容易发生。
与中间体3IM1和3IM1a的构型差异类似,过渡态3TS1和3TS1a的构型差别仍主要是O1―O2与S―H2的空间位置取向(3TS1中O1、O2、S和H2四原子所组成的二面角O1―O2―S―H2为90.8°,而3TS1a中二面角O1―O2―S―H2则为-85.8°)不同所致,过渡态3TS1与3TS1a的虚频分别为362i和365i cm-1,且其合振动方式均表现为H1原子沿HO2自由基中O2原子与HS自由基中S原子间的振动,证实3TS1和3TS1a确实是HO2中H原子迁移到HS中S原子上生成产物P1(H2S+3O2)的过渡态,此外,3TS1和3TS1a中即将断裂的H1―O2键与将要形成的S―H1键的键长变化率(δr[H1―O2]/δr[S―H1])均为0.13,表明3TS1和3TS1a均是类反应物过渡态,与Hammond35假设中放热反应过渡态具有类似反应物的几何构型一致。
图3绘出了路径Path 1和Path 1a中断键与成键的变化情况。如图3所示,路径Path 1主要发生在反应坐标s=-0.45-1.55amu1/2∙Bohr的反应区域。路径Path 1a也发生在类似的反应区间(s=-0.40-1.55amu1/2∙Bohr),表明路径Path 1和Path 1a所对应的能垒较窄,且该反应过程中存在着显著的隧道效应。因此计算路径Path 1和Path 1a的速率常数时,本文将考虑隧道效应
在有机硫化物CH3SO、CH3S以及C2H5S与HO2反应36-38中,均找到了类似与通道R2的途径。对于通道R2而言,HS自由基中的S原子抽取HO2中的O1部分,经过渡态3TS2克服61.25kJ∙mol-1的能垒最终生成产物P2(HSO+OH)。比较HO2与3TS2构型可以看出,HO2中欲断裂的O1―O2键长从0.1325nm拉伸到了3TS2中的0.1519 nm,新形成的O1―S键长在3TS2中缩短到了0.1910nm,在HSO中继续缩短至0.1511 nm。此反应过程是HS自由基抽氧反应,涉及一个O―O键断裂与一个O―S键生成,由于O―S键的键能远大于O―O键,因此可以预测此反应的能垒较高。从表1和图2可知,过渡态3TS2相对于反应物的能量比通道R1中过渡态3TS1和3TS1a相对于反应物的能量高出了大约66kJ∙mol-1,表明从动力学角度而言,通道R2不易发生。但与通道R1类似,通道R2也为放热反应,其反应吉布斯自由能值为-131.54kJ∙mol-1,表明从热力学角度而言通道R2却是有利进行的。因此加入适当的反应催化剂,改变通道R2的反应机理,便可以促使该反应通道的进行。
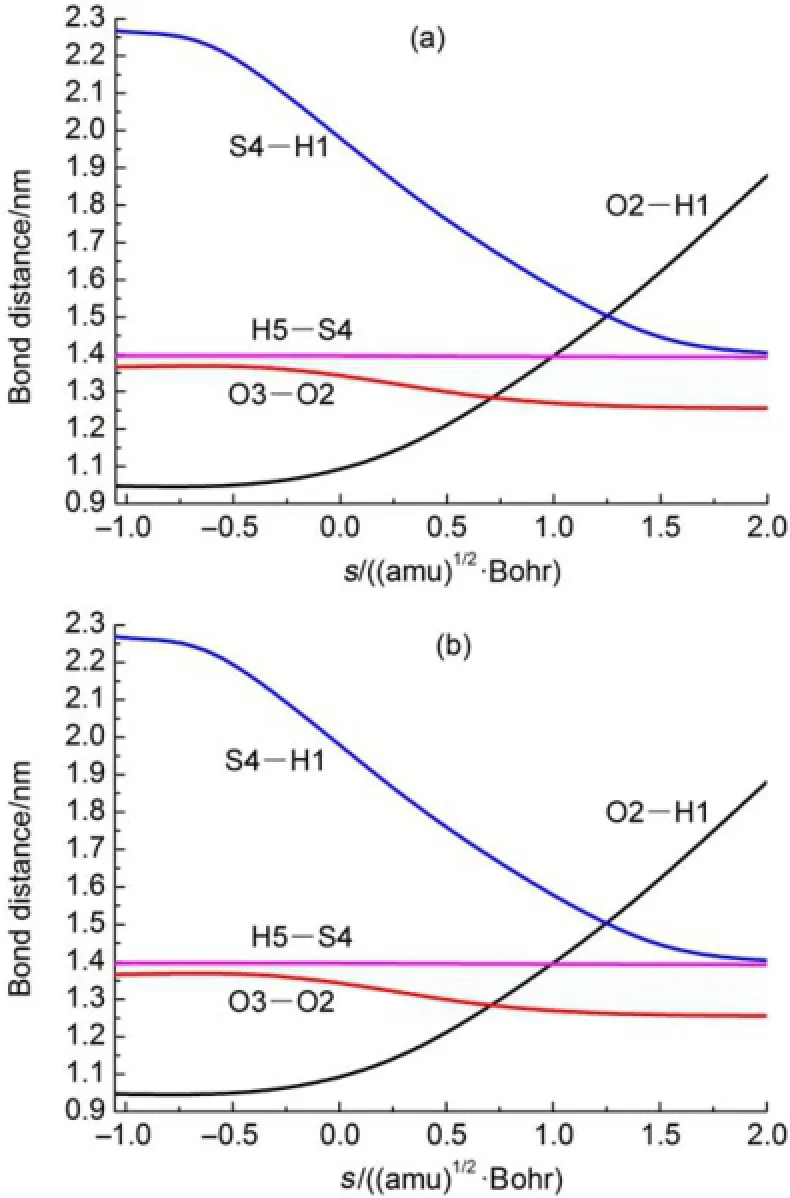
图3 在B3LYP/6-311+G(2df,2p)水平上Path 1(a)和Path 1a(b)速控步骤中键长随反应坐标(s)的变化Fig.3 Variationsof thebond distancesas function of reaction coord inate(s)about rate determ ining step of Path 1(a)and Path 1a(b)at B3LYP/6-311+G(2df,2p)level
3.1.2单重态HS+HO2反应势能面
在单重态反应势能面上找到了通道R5、R2、R7、R8和R9五条反应通道,分别生成产物P5(H2S+1O2)、P2(HSO+OH)、P7(1HSOOH)、P8 (HSOH+1O)和P9(HOOH+1S)。其中,通道R8和R9的能垒都很高(分别为150.01和140.53 kJ∙mol-1),且其产物P8和P9分别比反应物HS+HO2的能量高出195.61 kJ∙mol-1和79.24kJ∙mol-1。因此,从热力学和动力学角度考虑通道R8和R9是单重态反应势能面上的不利通道。此外对通道R2而言,与三重态路径Path 2类似,示意图1和图2所示的单重态路径Path 6和Path 6a同样可以生成产物P2(HSO+OH)。但与路径Path 2不同的是,其一,与三重态过渡态3TS2相比,单重态过渡态1TS6和1TS6a中的∠O2―O1―S较为平坦(1TS6和1TS6a中的∠O2―O1―S分别为178.9°和177.9°,而过渡态3TS2中∠O2―O1―S为143.8°),所受角张力较小;其二,路径Path 6和Path 6a在产物P2(HSO+OH)和过渡态(1TS6和1TS6a)之间形成中间体1IMF6(图1)。中间体1IMF6的稳定化能为182.98 kJ∙mol-1,其稳定性可能与该构型中较强的O―S(0.1646nm)键的存在有关。由图2和表1中的能量信息可知,经单重态过渡态1TS6(Path 6)和1TS6a(Path 6a)生成产物P2(HSO+OH)的能垒分别是187.12和198.74kJ∙mol-1,比三重态势能面上对应路径Path 2的能垒高出了125.87-137.49 kJ∙mol-1。基于此,此处仅对单重态势能面上较为优势的反应通道R5和R7进行论述。
与三重态反应通道R1类似,单重态反应通道R5(Path 5和Path 5a)也可以看成是HS中的S进攻HO2中的H,分别越过49.04和49.45kJ∙mol-1生成产物P5(H2S+1O2)。与路径Path 1和Path 1a(通道R1)不同,路径Path 5和Path 5a没有经过中间体而直接形成二面角O1―O2―S―H2和O1―O2―S―H2分别为-3.5°和166.7°的单重态过渡态1TS5和1TS5a。与过渡态3TS1与3TS1a的构型差别相似,单重态过渡态1TS5和1TS5a的差别仍在于O1―O2与S―H1排列取向不同,但与3TS1和3TS1a中的键角∠O2―H1―S相比,1TS5和1TS5a中的键角∠O2―H1―S较为弯曲(120.3°和119.8°)。在过渡态1TS5和1TS5a中即将形成的S―H1键的距离分别为0.1749和0.1759 nm,欲断裂的H1―O2键距离仅拉长到0.1087和0.1082 nm,具有明显的前过渡态特点。
在OH与HO2反应33中,也存在与通道R7类似的途径。与三重态HS+HO2反应不同,单重态反应通道R7可以认为是HS中的S原子与HO2O1自由基中非端基O2原子耦合的同时伴随着HO2O1自由基中端基(O1)原子抽取该自由基中H原子。从反应物R(HO2+HS)出发,通道R7直接经过渡态1TS7越过60.36kJ∙mol-1的能垒生成产物P7 (1HSO2H)。由图1和图2可以发现,由于HO2自由基端基O原子抽取该自由基中H原子比通道R5中SH自由基中的S原子抽取HO2自由基H原子要困难的多,因此从表1和图2所示的能量信息可知,在CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G (2df,2p)水平上通道R7的表观活化能比通道R5的表观活化能高出了11.32 kJ∙mol-1。由此可知,通道R5是HO2+SH反应单重态势能面上的主通道。但与三重态势能面上的主通道R1相比,通道R5的能垒高出了大约53.96kJ∙mol-1,表明单重态势能面上的主通道R5是整个反应体系中的次要通道。
3.2主通道速率常数计算
通过上述分析,通道R1是HO2+HS反应单、三重态势能面上的优势反应通道,因此本部分将对通道R1在200-800K温度范围内的速率常数进行计算。将通道R1中路径Path 1和Path 1a的反应平衡常数分别标记为和,速控步速率常数分别标记为和,反应速率常数分别标记为和,此外将通道R1的CVT/SCT速率常数标记为。表2列出了通道R1在CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上的以上速率常数信息。
从表2列出的路径Path 1和Path 1a的kCVT/SCT速率常数可知,在计算温度范围内,尽管路径Path 1a的速率常数总是大于路径Path 1的速率常数,但对应温度下仅比大了1.04-4.50倍。基于此在200-800K范围内Path 1和Path 1a的贡献均不可忽视。因此,下面对路径Path 1和Path 1a的微观动力学特征进行分析。图4给出了主路径Path 1和Path 1a的最小势能(VMEP(s))曲线、零点能(VZPE(s))曲线以及振动绝热基态势能()曲线曲线。
由图4可知,路径Path 1和Path 1a的零点能在鞍点区间附近有所降低,这使得VMEP(s)与的形状在该区间内存在微小差别,表明变分效应对路径Path 1和Path 1a的速率常数有所影响。为了清楚地了解变分效应和隧道效应对主路径Path 1和Path 1a中氢迁移动力学特征的影响,图5给出了200-800K温度范围内主路径Path 1和Path 1a的kTST、kCVT和kCVT/SCT速率常数随温度的变化情况。由图5可知,在计算温度范围内变分效应对Path 1和Path 1a的速率常数有一定影响。此外,隧道效应在较低温段对速率常数的影响显著。在200-800K温度区间内,主路径Path 1和Path 1a的速率常数kCVT/SCT可以分别拟合为如下三参数表达式:

表2 在CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上主通道R1中路径Path 1和Path 1a在200-800K温度范围内的速率常数信息Table2 Rate constants in formation of the Path 1 and Path 1a for ChannelR1 at the CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)level in the temperature range200-800K
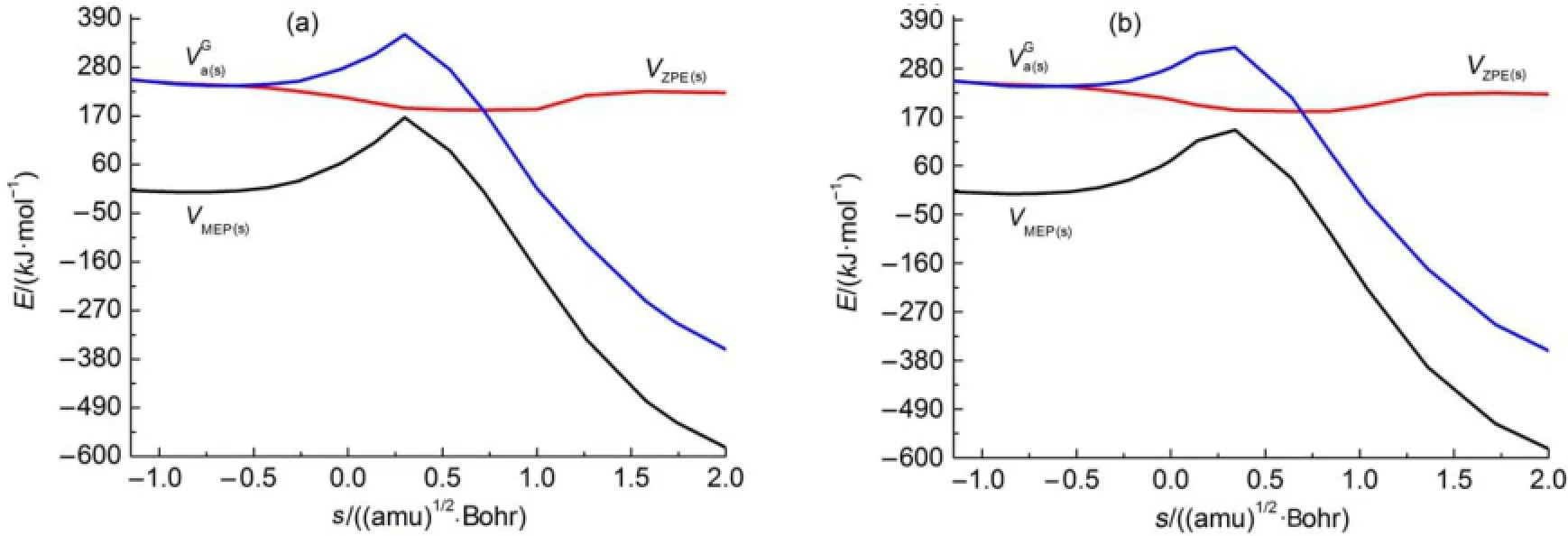
图4 Path 1(a)和Path 1a(b)控速步骤在CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上的最小势能(VMEP(s))、零点能(VZPE(s))和绝热势能曲线Fig.4Them inim um potentialenergy(V)and zero pointenergy(V),and vib rationally ad iabatic potentialenergyMEP(s)ZPE(s)curvesof ratedeterm ining step of Path 1(a)and Path 1a(b)at theCCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)level

图5 200-800K温度范围内CCSD(T)/6-311++G(3df,2pd)//B3LYP/6-311+G(2df,2p)水平上Path 1和Path 1a的表观速率常数(kTST、kCVT和kCVT/SCT)随温度的变化Fig.5Apparent rate constants(kTST,kCVT,and kCVT/SCT)for Path 1 and Path 1a calculated at the CCSD(T)/6-311++G(3df,2pd)// B3LYP/6-311+G(2df,2p)versus the recip rocalof tem perature over the range of200-800K
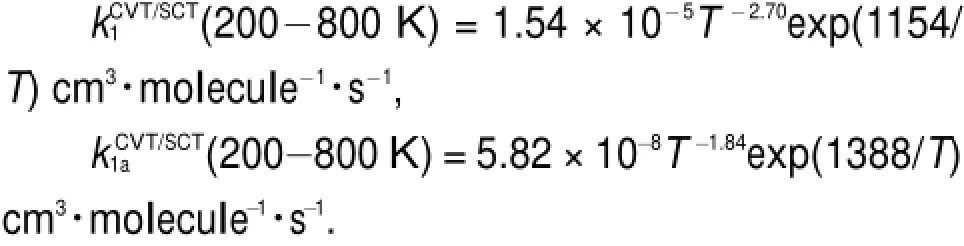
4 结论
通过对HO2+HS大气反应机理分析及速率常数计算,可得到如下结论。
(1)在大气环境中HO2+HS反应机理较为复杂,通过八条单、三重态通道产生H2S、3O2、HSO、OH、3O、HSOH、1O2、H2O2、1S、3S和HSOOH等物种,其中H2S和3O2是该反应的主要产物。
(2)主要产物H2S和3O2所在的三重态反应通道R1是标题反应的优势通道,在计算温度范围内该通道所包含的路径Path 1和Path 1a为竞争反应路径。
(3)在200-800K温度范围内,主路径Path 1和Path 1a的速率常数均随温度的升高而减小呈现出负温度系数效应,其CVT/SCT速率常数的三参数表达式分别为(200-800K)=1.54× 10-5T-2.70exp(1154/T)cm3∙molecule-1∙s-1和(200-800K)=5.82×10-8T-1.84exp(1388/T)cm3∙molecule-1∙s-1。
(4)HS中的S抽取HO2中的OH部分以及HO2端基氧原子抽取HS中的H是两种新的进攻方式,在OH+HO2反应体系中还未见报道。
Refe ren ces
(1)Farquhar,J.;Bao,H.;Thiemens,M.Science2000,289(5184), 756.doi:10.1126/science.289.5480.756
(2)Martínez,E.;Albaladejo,J.;Notario,A.;Jiménez,E.Atmos. Environ.2000,34(29),5295.doi:10.1016/S1352-2310(00) 00348-4
(3)Vandeputte,A.G.;Reyniers,M.F.;Marin,G.B.J.Phys. Chem.A 2010,114(39),10531.doi:10.1021/jp103357z
(4)W illiams,M.B.;Campuzano-Jost,P.;Hynes,A.J.;Pounds,A. J.J.Phys.Chem.A 2009,113(24),6697.doi:10.1021/ jp9010668
(5)Wang,W.;Xin,J.;Zhang,Y.;Wang,W.;Lu,Y.Int.J.Quantum Chem.2011,111(3),644.doi:10.1002/qua.22446
(6)Yan,J.;Yang,J.;Liu,Z.Environ.Sci.Technol.2005,39(13), 5043.doi:10.1021/es048398c
(7)Resende,S.M.;Ornellas,F.R.J.Phys.Chem.A 2000,104(51),11934.doi:10.1021/jp001751q
(8)Black,G.J.Chem.Phys.1984,80(3),1103.doi:10.1063/ 1.446838
(9)Atkinson,R.;Baulch,D.L.;Cox,R.A.;Crow ley,J.N.; Hampson,R.F.;Hynes,R.G.;Jenkin,M.E.;Rossi,M.J.; Troe,J.Atmos.Chem.Phys.2004,4(6),1461.doi:10.5194/ acp-4-1461-2004
(10)Herndon,S.C.;Froyd,K.D.;Lovejoy,E.R.;Ravishankara,A. R.J.Phys.Chem.A 1999,103(34),6778.doi:10.1021/ jp9911853
(11)Friedl,R.R.;Brune,W.H.;Anderson,J.G.J.Phys.Chem. 1985,89(25),5505.doi:10.1021/j100271a038
(12)Nesbitt,D.J.;Leone,S.R.J.Chem.Phys.1980,72(3),1722. doi:10.1063/1.439284
(13)Domagal-Goldman,S.D.;Meadows,V.S.;Claire,M.W. Astrobiology2011,11(5),419.doi:10.1089/ast.2010.0509
(14)Shum,L.G.S.;Benson,S.W.Int.J.Chem.Kinet.1985,17 (7),749.doi:10.1002/kin.550170705
(15)Imai,N.;Toyama,O.Bull.Chem.Soc.Jpn.1961,34(3),328. doi:10.1246/bcsj.34.328
(16)Amphlett,J.C.;Whittle,E.Trans.Faraday Soc.1967,63, 2695.doi:10.1039/TF9676302695
(17)Perner,V.D.;Franken,T.Ber.Bunsenges.Phys.Chem.1969, 73(8-9),897.doi:10.1002/bbpc.19690730830
(18)Long,B.;Zhang,W.J.;Tan,X.F.;Long,Z.W.;Wang,Y.B.; Ren,D.S.J.Phys.Chem.A 2011,115(8),1350.doi:10.1021/ jp107550w
(19)Allodi,M.A.;Dunn,M.E.;Livada,J.;Kirschner,K.N.; Shields,G.C.J.Phys.Chem.A 2006,110(49),13283.doi: 10.1021/jp064468l
(20)Zhou,Y.Z.;Zhang,S.W.;Li,Q.S.Chem.J.Chin.Univ.2006, 27(8),1496.[周玉芝,张绍文,李前树.高等学校化学学报, 2006,27(8),1496.]
(21)Gonzalez,C.;Schlegel,H.B.J.Chem.Phys.1989,90(4), 2154.doi:10.1063/1.456010
(22)Lee,Y.S.;Kucharski,S.A.;Bartlett,R.J.J.Chem.Phys. 1984,81(12),5906.doi:10.1063/1.447591
(23)Liu,Z.R.;Xu,B.E.;Zeng,Y.L.;Li,X.Y.;Meng,L.P.;Sun, Z.;Zhang,X.Y.;Zhang,P.Acta Chim.Sin.2011,69(17), 1957.[刘占荣,许保恩,曾艳丽,李晓艳,孟令鹏,孙政,张雪英,张萍.化学学报,2011,69(17),1957.]
(24)Xu,Q.;Wang,R.;Zhang,T.L.;Zhang,H.L.;Wang,Z.Y.; Wang,Z.Q.Chem.J.Chin.Univ.2014,35(10),2191.[许琼,王睿,张田雷,张浩林,王志银,王竹青.高等学校化学学报,2014,35(10),2191.]doi:10.7503/cjcu20140310
(25)Frisch,M.J.;Trucks,G.W.;Pople,J.A.;etal.Gaussian 09, Revision A.01;Gaussian Inc.:Pittsburgh,PA,2009.
(26)Zhang,S.W.;Truong,N.T.VKLab,version 1.0;University of U tah,Salt Lake City,USA,2001.
(27)Garrett,B.C.;Truhlar,D.G.;Grev,R.S.;Magnuson,A.W. J.Phys.Chem.1980,84(13),1730.doi:10.1021/j100450a013
(28)Liu,Y.P.;Lynch,G.C.;Truong,T.N.;Lu,D.H.;Truhlar,D. G.;Garrett,B.C.J.Am.Chem.Soc.1993,115(6),2408.doi: 10.1021/ja00059a041
(29)Anglada,J.M.;Domingo,V.M.J.Phys.Chem.A 2005,109 (47),10786.doi:10.1021/jp054018d
(30)Si,W.J.;Zhuo,S.P.;Ju,G.Z.Acta Phys.-Chim.Sin.2003,19 (10),974.[司维江,禚淑萍,居冠之.物理化学学报,2003,19 (10),974.]doi:10.3866/PKU.WHXB20031019
(31)From theNIST chemistry webbook,http://webbook.nist.gov/ chem istry.
(32)Gonzalez,C.;Theisen,J.;Zhu,L.;Schlegel,H.B.;Hase,W. L.;Kaiser,E.W.J.Phys.Chem.1991,95(18),6784.doi: 10.1021/j100171a010
(33)Gonzalez,C.;Theisen,J.;Schlegel,H.B.;HaseW.L.;Kaiser, E.W.J.Phys.Chem.1992,96(4),1767.doi:10.1021/ j100183a051
(34)Zhang,T.L.;Wang,W.L.;Li,C.Y.;Du,Y.M.;Lv,J.RSC Adv.2013,3(20),7381.doi:10.1039/c3ra40341f
(35)Hammond,G.S.J.Am.Chem.Soc.1955,77(2),334.doi: 10.1021/ja01607a027
(36)Lu,Y.X.;Wang,W.L.;Wang,W.N.;Liu,Y.Y.;Zhang,Y. Acta Chim.Sin.2010,68(13),1253.[卢彦霞,王文亮,王渭娜,刘英英,张越.化学学报,2010,68(13),1253.]
(37)Liu,Y.;Wang,W.;Zhang,T.;Cao,J.;Wang,W.;Zhang,Y. Comput.Theor.Chem.2011,964(1),169.doi:10.1016/j. comptc.2010.12.017
(38)Zhang,Y.;Zhang,W.;Zhang,T.;Tian,W.;Wang,W.Comput. Theor.Chem.2012,994,65.doi:10.1016/j. comptc.2012.06.016
Theoretical Study on the Atmospheric Reaction of HS with HO2: Mechanism and Rate Constants of the Major Channel
ZHANG Tian-Lei1,*YANG Chen1FENG Xu-Kai1WANG Zhu-Qing2WANG Rui1LIU Qiu-Li1ZHANG Peng1WANGWen-Liang3,*
(1ShaanxiProvince Key Laboratory ofCatalytic Fundamental&Application,SchoolofChemical&EnvironmentScience, ShaanxiUniversity ofTechnology,Hanzhong 723001,ShaanxiProvince,P.R.China;2Shandong Provincial Key Laboratory of Ocean EnvironmentMonitoring Technology,Shandong Academy ofSciences Institute ofOceanographic Instrumentation, Qingdao 266001,Shandong Province,P.R.China;3Key Laboratory forMacromo lecular Science ofShaanxiProvince, SchoolofChemistry and Chemical Engineering,ShaanxiNormalUniversity,Xi′an 710062,P.R.China)
Themechanism for the biradical reaction ofHSwith HO2is investigated at the CCSD(T)/6-311++ G(3df,2pd)//B3LYP/6-311+G(2df,2p)levelon both the sing letand trip letpotentialenergy surfaces,along w ith rate constant calculations of themajor channel.The results show that there are eight reaction channels involved in the HS+HO2reaction system.Themajor channelR1 of the title reaction occurs on the tripletpotentialenergy surfaces,and includes two pathways:Path 1(R→3IM1→3TS1→P1(3O2+H2S))and Path 1a(R→3IM1a→3TS1a→P1(3O2+H2S)).The rate constants kTST,kCVT,and kCVT/SCTof Paths 1 and 1a for Channel R1 were evaluated using classical transition state theory(TST)and the canonicalvariational transition state theory(CVT),in which the sma ll-curva ture tunneling correction was included.The calcula ted results show tha t kTST,kCVT,and kCVT/SCTof these two pathways decrease with rising tem perature w ithin the temperature range of 200-800K.The variationaleffectwas notnegligible in the en tire p rocess of Path 1 and Path 1a,at the sam e time,the tunneling effectwas considerab le at lower tem perature.The fitted three-parameterexpressions o f kCVT/SCTforPaths 1 and 1a are(200-800K)=1.54×10-5T-2.70exp(1154/T)cm3∙mo lecule-1∙s-1and(200-800K)=5.82×10-8T-1.84exp(1388/T)cm3∙m olecu le-1∙s-1,respectively.
November20,2015;Revised:December28,2015;Published onWeb:December30,2015.
HS;HO2;Potentia lenergy surface;Reactionmechanism;Rate constant
O643.1
10.3866/PKU.WHXB201512303
*Corresponding authors.ZHANGTian-Lei,Email:ztianlei88@163.com.WANGWen-Liang,Email:w lwang@snnu.edu.cn;Tel:+86-916-2641083.
Theprojectwas supported by the NationalNaturalScience Foundation ofChina(21473108,21207081)and FundsofResearch Programsof Shaanxi University of Technology,China(SLGQD13(2)-3,SLGQD13(2)-4).
国家自然科学基金(21473108,21207081)及陕西理工学院科研计划项目(SLGQD13(2)-3,SLGQD13(2)-4)资助©Editorialofficeof Acta Physico-Chimica Sinica
