Actinonin与肽脱甲酰基酶作用模式的分子动力学模拟研究
2016-09-07杰徐州医学院江苏省新药研究与临床药学重点实验室江苏徐州221004
高 剑 王 涛 孙 杰 牟 杰徐州医学院 江苏省新药研究与临床药学重点实验室,江苏徐州 221004
Actinonin与肽脱甲酰基酶作用模式的分子动力学模拟研究
高剑王涛孙杰牟杰
徐州医学院江苏省新药研究与临床药学重点实验室,江苏徐州221004
目的 研究actinonin与脱甲酰基酶的相互作用模式,阐述基于actinonin进行特异性人肽脱甲酰基酶(HsPDF)抑制剂设计思路。 方法使用分子动力学模拟和MM/GBSA自由能计算等方法来研究各种肽脱甲酰基酶(PDF)与actinonin的相互作用模式,定量描述PDF关键残基与actinonin的结合自由能。结果 在与actinonin结合方面,HsPDF作为PDF1A的代表,与PDF1B和PDF2比较,存在3个明显差异:与HsPDF的motif 1相互作用较强,与motif 2较弱,而与PDF1B和PDF2的motif 1相互作用较弱,而与motif 2相互作用较强;与HsPDF的Leu131和Met145相互作用较强,而这些相互作用在PDF1B和PDF2是不存在的;与HsPDF的Trp207存在很强的结合自由能,而与PDF1B和PDF2在相同位置的残基相互作用较弱。结论利用分子动力学模拟的方法,研究actinonin与PDF的相互作用模式,阐述HsPDF与其他类型PDF在活性位点的差异,为基于actinonin的特异性HsPDF抑制剂的设计提出了思路。
分子动力学模拟;肽脱甲酰基酶;Actinonin;抗肿瘤;抗菌
肽脱甲酰基酶(peptide deformylase,PDF)是含铁金属蛋白酶,功能是脱去新生成的N-甲酰甲硫氨酸多肽的N-甲酰基,是蛋白质合成过程中必不可少的一种酶[1-2]。PDF有3种类型:PDF1、PDF2、PDF3,是无脱甲基化功能,其中PDF1又分为两个亚型,PDF1A(如HsPDF)和PDF1B(细菌PDF)[3-5]。原核细胞中的PDF已被当作极具潜力的抗菌靶标,且大量PDF抑制剂已经被报道[6-10]。然而肽脱甲酰基酶(PDF-like)也存在于真核细胞中,如寄生虫、植物、甚至哺乳动物的人类[4,11-12]。HsPDF分布于人的线粒体中,同样具有对底物多肽的脱甲基化的功能[13-16],而这对于线粒体中DNA所编码蛋白的积聚和装配至关重要[17]。抑制HsPDF将降低线粒体的呼吸功能及相应的能量产生,最终导致细胞死亡[17]。最新研究也证实HsPDF在乳腺癌、结肠癌和肺癌中高表达,抑制HsPDF将明显降低癌细胞的增殖[18]。林超等[19]研究表明,HsPDF基因在Hela、MDA-MB-231、K562等肿瘤细胞中高表达,而在人正常肺组织中低表达(P<0.01),Actinonin(图1)是首个天然来源的细菌PDF抑制剂[7],被证实是有潜力的HsPDF抑制剂 (IC50为43 nmol/L)[20-21]。因此,HsPDF是极具潜力的新型抗肿瘤靶标。然而以actinonin为代表的HsPDF抑制剂由于其可以同时抑制人体内的细菌群的PDF蛋白酶而制约着此类抑制剂的发展[22],亟待特异性HsPDF抑制剂的设计与开发。
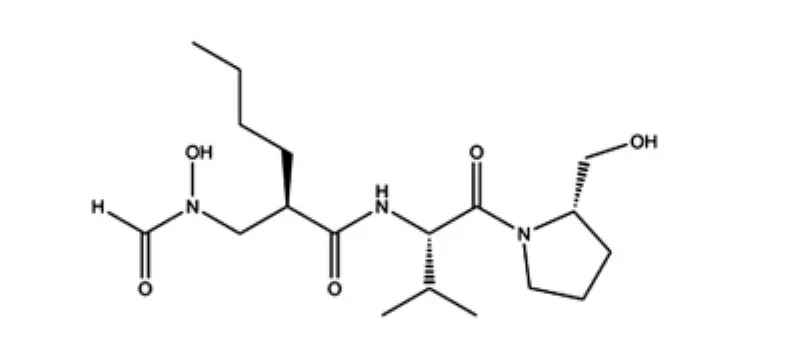
图1 PDF抑制剂actinonin的结构
组合使用分子动力学(molecular dynamics,MD)模拟和结合自由能计算的方法已经成熟应用于定量描述蛋白-蛋白和蛋白-小分子相互作用模式中[23-29]。为了阐述各个类型PDF蛋白酶抑制剂结合位点的特性,设计了7个PDF(1个PDF1A、3个PDF1B和3个PDF2)与其抑制剂actinonin形成复合物的体系,利用分子动力学模拟和结合自由能计算等方法,定量描述了多种类型PDF蛋白酶抑制剂结合位点的共性与差异性。7个体系是由1个PDF1A(HsPDF-actinonin)、3个PDF1B[包括聚球藻类噬菌体S-SSM7 PDF-actinonin(简称SpPDF-actinonin)、幽门螺杆菌PDF-actinonin(简称HpPDF-actinonin)和拟南芥PDF-actinonin(简称At PDF-actinonin)]和3个PDF2[包括金黄色葡萄球菌PDF-actinonin(简称SaPDF-actinonin)、嗜热脂肪芽孢杆菌PDF-actinonin(简称BsPDF-actinonin)和蜡样芽孢杆菌PDF-actinonin(简称BcPDF-actinonin)]组成的。
1 资料与方法
1.1模拟体系构建
以上提及的7个复合物体系均来自于蛋白质数据库(PDB)[30],相应晶体结构PDB号分别为HsPDF-actinonin:3G5K[22]、SpPDF-actinonin:3UWB[31]、HpPDF-actinonin:4E9B、AtPDF-actinonin:3M6P[32]、SaPDF-actinonin:1Q1Y[33]、BsPDF-actinonin:1LQY[3]和 BcPDF-actinonin:2OKL[34]。7个晶体结构均含有抑制剂actinonin。

图2 7种PDF蛋白酶的序列比对
1.2分子动力学(MD)模拟和相应的分子力场
7个复合物体系的能量优化和分子动力学模拟均采用Amber12分子动力学程序包,PDF酶的参数采用FF03力场,actinonin的参数采用GAFF力场。每个体系置于TIP3P水分子的立方体盒中,并保证复合物的四周均包上厚度为12的水层。为使各个体系呈现电中性,适量的Na+以替换相同数量水分子的形式加入体系中。每个体系先利用sander模块进行能量优化。之后对每个体系进行60 ps的逐步升温过程(0~300 K),体系处于NVT的系综中。每个体系的MD模拟均在恒温300 K下运行20 ns,步长是2 fs。
1.3MM/GBSA结合自由能计算方法
PDF与actinonin的结合自由能的计算采用MM/ GBSA方法,由MM_PBSA和Nmode模块来完成,计算公式如下:
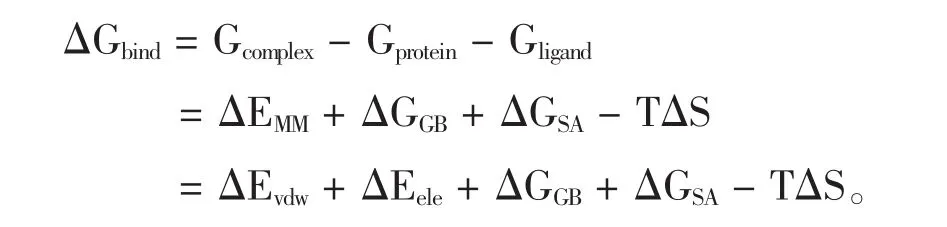
其中ΔEMM为气相分子间相互作用能,包括范德华(ΔEvdw)和静电相互作用(ΔEele),ΔGGB和 ΔGSA分别代表了极性溶剂化和非极性溶剂化效应,-TΔS代表了温度的熵变。每个体系的结合自由能计算是基于最后10 ns的轨迹中均匀选取的400个构象得到的。
1.4MM/GBSA自由能分解
为了定量计算受体PDF活性位点残基与抑制剂的相互作用能,采用Mm_Pbsa模块进行MM/GBSA自由能分解。单个残基与抑制剂的结合自由能同样包含3个部分:范德华、静电相互作用和溶剂化贡献。
2 结果
2.1MD模拟轨迹分析
为了考察每个体系在分子动力学模拟过程中蛋白结构构象的稳定性,计算了蛋白主链骨架原子相对于初始构象的均方根偏差(RMSD),如图3所示。所有7个体系在较短的时间内就达到稳态,RMSD值均在1.2左右。7个体系在10 ns之后均达到稳态,从最后10 ns的轨迹中选取400个构象进行结合自由能计算是合理的。
2.2MM/GBSA结合自由能计算
采用MM/GBSA计算PDF与actinonin的结合自由能,结果列于表1。actinonin与HsPDF、SpPDF、HpPDF、AtPDF、BcPDF、SaPDF和BsPDF的结合自由能分别为-115.81、-123.31、-150.17、-130.40、-119.42、-126.44 kJ/mol和-127.28 kJ/mol。每个体系计算得到的结合自由能与实验值(p IC50)的相关系数较高(r2= 0.81)。范德华作用对抑制剂与PDF蛋白酶结合起关键作用,总的非极性相互作用(ΔEvdw+ΔGSA,简称为ΔG1)的贡献明显高于极性相互作用(ΔEele+ΔGGB,简称为ΔG2)的贡献。
2.3MM/GBSA自由能分解
各种PDF氨基酸序列同源性不高,但大多存在3个保守的氨基酸片段:GXGXAAXQ(motif 1)、EGCLS(motif 2)和HEXXH(motif 3)(X代表任意残基)。MM/ GBSA自由能分解得到了单个残基与抑制剂定量的相互作用能(图4)也显示出actinonin与7个不同的PDF起关键相互作用的残基大都归属于上面3个片段。actinonin在 3个 PDF1B类型(SpPDF、HpPDF和AtPDF)和3个PDF2类型(BcPDF、SaPDF和BsPDF)中的结合模式较为相似(图4)。actinonin与PDF1A类型(HsPDF)的结合模式与之前6个体系比较存在一定差异,如HsPDF中的L131和M145。另外,HsPDF 中207位的氨基酸是色氨酸,而该位置在其他类型PDF酶中多是亮氨酸。
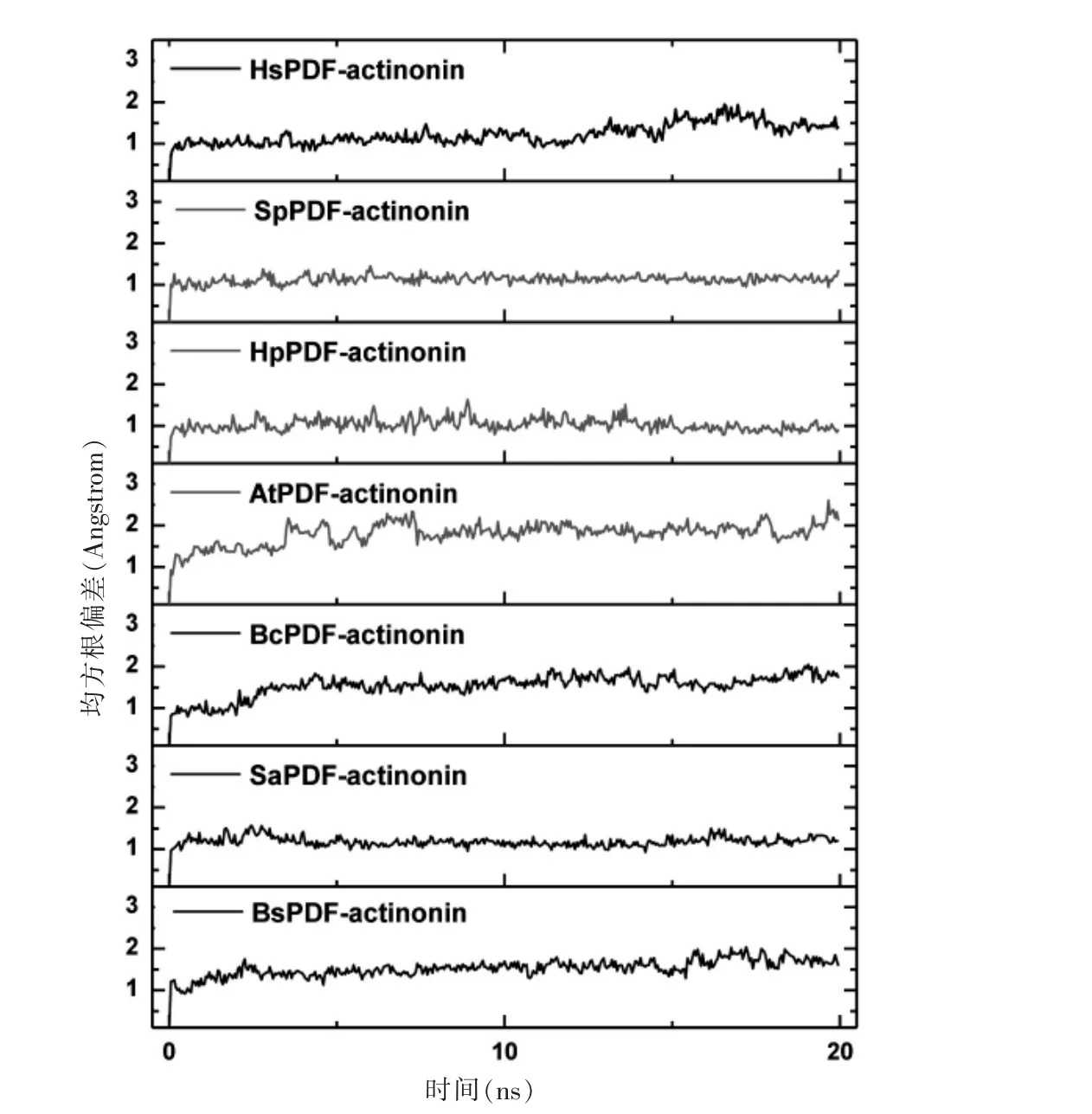
图3 MD模拟过程中蛋白主链骨架原子相对于初始构象的均方根偏差
为了进一步考察actinonin与不同类型PDF酶相互作用的共性与差异性,3个保守片段及其他重要残基在结合自由能中的贡献结果列于表2。actinonin与HsPDF的motif 1的结合自由能(-29.45 kJ/mol)强于与其他类型的motif1(均在-25 kJ/mol附近),与motif2的结合自由能(-19.62 kJ/mol)弱于PDF2型,更低于PDF1B型,与motif 3的结合自由能与其他两种类型比较没有明显差异(图4)。另外,actinonin与HsPDF 的Leu131和Met145有-3.18 kJ/mol和-3.14 kJ/mol的结合自由能,而与其他两种类型PDF在相同位置的残基相互作用为零,这些结果也在图5(封三)中呈现出。最后,actinonin与HsPDF的Trp207有极强的结合自由能(-11.72 kJ/mol),而与其他类型PDF相互作用较弱。
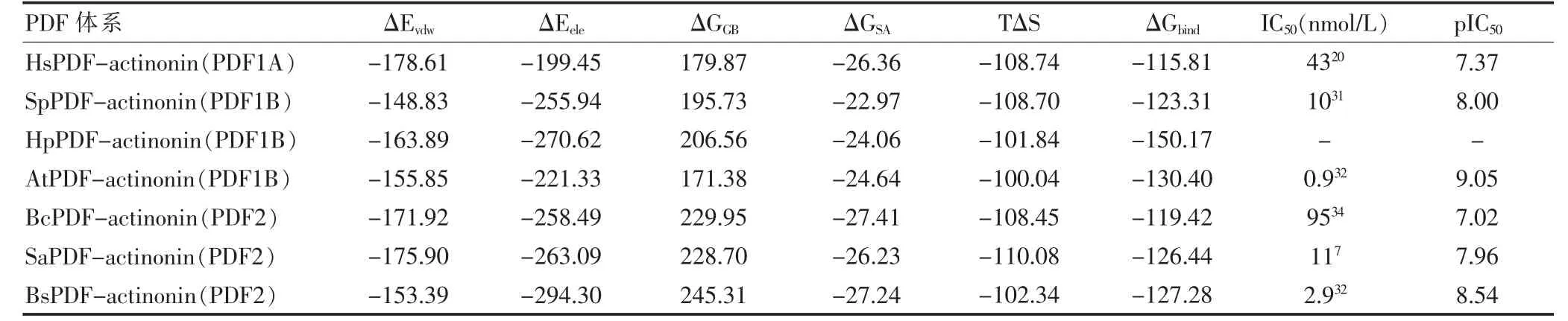
表1 7个体系各自的结合自由能及各能量项对结合自由能的贡献
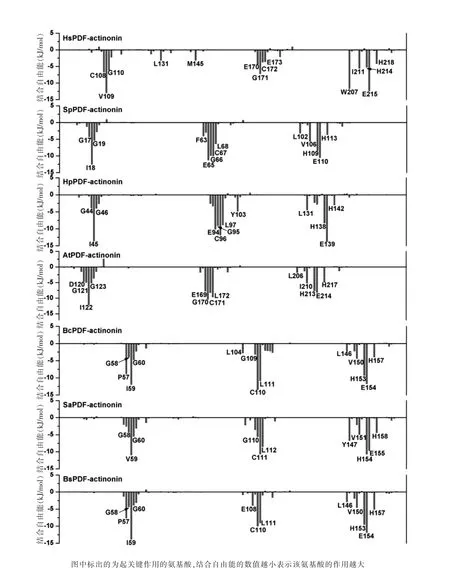
图4 各种PDF与actinonin的相互作用

表2 actinonin与关键氨基酸片段总的结合自由能(kJ/mol)
3 讨论
PDF可以作为抗菌和抗肿瘤的双重靶标。PDF作为抗菌靶标研究较早并且已经有几个小分子化合物进入临床试验,而基于HsPDF进行抗肿瘤的研究起步较晚,尚无良好的HsPDF抑制剂。actinonin作为高效的细菌PDF抑制剂同时也显示出较强的HsPDF抑制活性,但其同时抑制多种PDF酶而限制了HsPDF抑制剂的发展。设计高特异性的HsPDF抑制剂将明显降低其对其他类型PDF的抑制活性而具有低毒副作用。为此,利用分子动力学模拟和结合自由能计算来研究actinonin与7个PDF的相互作用,通过定量考察actinonin与PDF活性位点残基的相互作用能来揭示actinonin结合于不同PDF时的差异:actinonin 与HsPDF的motif 1相互作用较强,与motif 2较弱。actinonin与PDF1B和PDF2的motif 1相互作用较弱,而与motif 2相互作用较强。同时,actinonin与HsPDF的Leu131和Met145为主的Helix 2和Helix 3的相互作用较强,而这些相互作用在PDF1B和PDF2是不存在的。最后,actinonin与HsPDF的Trp207有较强的结合自由能,而与PDF1B和PDF2在相同位置的残基相互作用则较弱。这些信息将为今后基于HsPDF进行抗肿瘤药物的发展提供理论指导。
[1]Leeds JA,Dean CR.Peptide deformylase as an antibacterial target:a critical assessment[J].Current Opinion in Pharmacology,2006,6(5):445-452.
[2]Rajagopalan PTR,Yu XC,Pei D.Peptide deformylase:a new type of mononuclear iron protein[J].Journal of the American Chemical Society,1997,119(50):12418-12419.
[3]Guilloteau JP,Mathieu M,Giglione C,et al.The crystal structures of four peptide deformylases bound to the antibiotic actinonin reveal two distinct types:a platform for the structure-based design of antibacterial agents[J].J Mol Biol,2002,320(5):951-962.
[4]Giglione C,Pierre M,Meinnel T.Peptide deformylase as a target for new generation,broad spectrum antimicrobial agents[J].MolMicrobiol,2000,36(6):1197-1205.
[5]Giglione C,Meinnel T.Peptide deformylase as an emerging target for antiparasitic agents[J].Expert Opin Ther Targets,2001,5(1):41-57.
[6]Meinnel T,Patiny L,Ragusa S,et al.Design and synthesis of substrate analogue inhibitors of peptide deformylase[J]. Biochemistry,1999,38(14):4287-4295.
[7]Chen DZ,Patel DV,Hackbarth CJ,et al.Actinonin,a naturally occurring antibacterial agent,is a potent deformylase inhibitor[J].Biochemistry,2000,39(6):1256-1262.
[8]Hu X,Nguyen KT,Verlinde CL,et al.Structure-based design of a macrocyclic inhibitor for peptide deformylase[J]. Journal of Medicinal Chemistry,2003,46(18):3771-3774.
[9]Lofland D,Difuntorum S,Waller A,et al.In vitro antibacterial activity of the peptide deformylase inhibitor BB-83698[J].Journal of Antimicrobial Chemotherapy,2004,53 (4):664-668.
[10]O'DwyerK,Hackel M,Hightower S,et al.Comparativeanalysis of the antibacterial activity of a novel peptide deformylase inhibitor,GSK1322322[J].Antimicrob Agents Chemother,2013,57(5):2333-2342.
[11]Giglione C,Serero A,Pierre M,et al.Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms[J].EMBO J,2000,19(21):5916-5929.
[12]Meinnel T.Peptide deformylase of eukaryotic protists:a target for new antiparasitic agents?[J].Parasitology Today,2000,16(4):165-168.
[13]Spencer AC,Spremulli LL.Interaction of mitochondrialinitiation factor 2 with mitochondrial fMet-tRNA[J]. Nucleic Acids Research,2004,32(18):5464-5470.
[14]Liao HX,Spremulli LL.Identification and initial characterization of translational initiation factor 2 from bovine mitochondria[J].Journal of Biological Chemistry,1990,265 (23):13618-13622.
[15]Pereira-Castro I,Costa LTd,Amorim A,et al.Transcriptional regulation of the human mitochondrial peptide deformylase(PDF)[J].Biochemical and Biophysical Research Communications,2012,421(4):825-831.
[16]Liu CC,Liu BG,Yang ZW,et al.Genome-wide identification and in silico analysis of poplar peptide deformylases[J].International Journal of Molecular Sciences,2012,13(4):5112-5124.
[17]Escobar-Alvarez S,Gardner J,Sheth A,et al.Inhibition of human peptide deformylase disrupts mitochondrial function[J].Mol Cell Biol,2010,30(21):5099-5109.
[18]Randhawa H,Chikara S,Gehring D,et al.Overexpression of peptide deformylase in breast,colon,and lung cancers[J]. BMC Cancer,2013,13(1):1-7.
[19]林超,罗学刚,邢莹莹,等.人肽脱甲酰基酶基因pdf与肿瘤细胞生长的相关性[J].中国药科大学学报,2006,37(5):474-478.
[20]Lee MD,Antczak C,Li Y,et al.A new human peptide deformylase inhibitable by actinonin[J].Biochemical and Biophysical Research Communications,2003,312(2):309-315.
[21]Han JH,Choi YS,Kim WJ,et al.Codon optimization enhancesprotein expression of human peptide deformylase in E.coli[J].Protein Expression and Purification,2010,70 (2):224-230.
[22]Escobar-Alvarez S,Goldgur Y,Yang G,et al.Structure and activity of human mitochondrial peptide deformylase,a novel cancer target[J].JMol Biol,2009,387(5):1211-1228.
[23]Gao J,Wang L,Kang SG,et al.Size-dependent impact of CNTs on dynamic properties of calmodulin[J].Nanoscale,2014,6(21):12828-12837.
[24]Hou T,Zhang W,Case DA,et al.Characterization of domain-peptide interaction interface:a case study on the Amphiphysin-1 SH3 domain[J].Journal of Molecular Biology,2008,376(4):1201-1214.
[25]Hou T,McLaughlin W,Lu B,et al.Prediction of binding affinities between the human Amphiphysin-1 SH3 domain and its peptide ligands using homology modeling,molecular dynamics and molecular field analysis[J].Journal of Proteome Research,2005,5(1):32-43.
[26]Hou T,Wang J,Li Y,et al.assessing the performance of the MM/PBSA and MM/GBSA methods.1.The accuracy of binding free energy calculations based on molecular dynamics simulations[J].Journal of Chemical Information and Modeling,2010,51(1):69-82.
[27]崔巍,张怀,计明娟.新型二氟甲基磷酸类酪氨酸蛋白磷酸酯酶1B抑制剂的分子动力学模拟和结合自由能计算[J].物理化学学报,2009,25(4):668-676.
[28]侯廷军,徐筱杰.基于分子动力学模拟和连续介质模型的自由能计算方法[J].化学进展,2004,16(2):153-158.
[29]罗芳,高剑,成元华,等.葡萄糖苷酶抑制剂作用机理的分子动力学模拟和自由能计算[J].物理化学学报,2012,9(9):2191-2201.
[30]Berman HM,Westbrook J,Feng Z,et al.The protein data bank[J].Nucleic Acids Res,2000,28(3):235-242.
[31]Frank JA,Lorimer D,Youle M,et al.Structure and function of a cyanophage-encoded peptide deformylase[J]. ISME J,2013,7(6):1150-1160.
[32]Fieulaine S,Boularot A,Artaud I,et al.Trapping conformational states along ligand-binding dynamics of peptide deformylase:the impact of induced fit on enzyme catalysis[J].PLoS Biol,2011,9(5):211-218.
[33]Yoon HJ,Kim HL,Lee SK,et al.Crystal structure of peptide deformylase from Staphylococcus aureus in complex with actinonin,a naturally occurring antibacterial agent[J]. Proteins,2004,57(3):639-642.
[34]Park JK,Kim KH,Moon JH,et al.Characterization of peptide deformylase 2 from B cereus[J].J Biochem Mol Biol,2007,40(6):1050-1057.
Molecular dynamics simulation on the molecular interactions of peptide deformylase and actinonin
GAO JianWANG Tao SUN Jie MOU Jie
Xuzhou Medical CollegeJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy,Jiangsu Province,Xuzhou221004,China
Objective To investigate the binding modes between actinonin and peptide deformylases and to characterize the informations about how to design the specific HsPDF inhibitor.Methods The combination of molecular dynamics simulation and MM/GBSA free energy calculation was employed to study the interactions between several PDF enzymes and actinonin.The detailed interactions of each residues of protein and actinonin were calculated by MM/GBSA free energy decomposition.Results Three important differences in actinonin binding to PDFs were obtained as followed: actinonin had stronger binding interaction with the motif 1 of HsPDF than the motif 2,while actinonin had lower binding interaction with the motif 1 than the motif 2 for PDF1A and PDF2.Moreover,actinonin had stronger binding affinities with Leu131 and Met145 of HsPDF,but had no interaction with the corresponding ones of PDF1A and PDF2.In addition,actinonin interacted strongly with Trp207 of HsPDF but weakly with the corresponding residue of PDF1A and PDF2.Conclusion By using the method of molecular dynamics simulation,the interaction of actinonin and PDFmodel are studied,differences of HsPDF and PDF in the active site with other types are expounded,based on the design thinking of specific HsPDF inhibitors of actinonin is proposed.
Molecular dynamics simulation;Peptide deformylase;Actinonin;Anticancer;Antibacterial
R75
A
1673-7210(2016)01(c)-0021-06
2015-09-22本文编辑:任念)
江苏省青年科学基金项目(BK20140225);徐州医学院优秀人才科研启动基金资助项目(D2014008)。
