PC12细胞氧糖剥夺模型细胞自噬与损伤的关系及黄芪甲苷的保护作用研究
2016-09-02黄小平李静娴杨筱倩刘晓丹邓常清
黄小平 李静娴 杨筱倩 丁 煌 唐 标 刘晓丹 邓常清
PC12细胞氧糖剥夺模型细胞自噬与损伤的关系及黄芪甲苷的保护作用研究
黄小平李静娴杨筱倩丁煌唐标刘晓丹邓常清
(湖南中医药大学分子病理实验室,中西医结合心脑疾病防治湖南省重点实验室,细胞生物学与分子技术湖南省高校重点实验室,长沙,410208)
目的:研究PC12细胞OGD/R后自噬与损伤的经时性变化以及黄芪甲苷的干预效应。方法:探讨PC12细胞OGD/ R后不同时间自噬的变化及与细胞损伤的关系,初步确定细胞发生自噬性损伤的时间点,再制作PC12细胞OGD/R自噬性损伤模型,研究黄芪甲苷抗自噬性损伤的作用。结果:复糖复氧6~36h后,细胞存活率降低、LDH漏出率增加;用3-MA预处理后,可使复糖复氧6~12h细胞存活率降低、LDH漏出率增加,复糖复氧后24~36h相反。激光共聚焦和western-blot检测显示,复糖复氧后6hLC3、LC3-Ⅱ/LC3-Ⅰ比值增加,至24h达高峰;p62蛋白表达随再复糖复氧时间的延长逐渐降低。黄芪甲苷对OGD 2h复糖复氧24h的PC12细胞自噬具有显著抑制作用,且呈剂量依赖性。结论:PC12在在OGD复糖复氧6~12h,自噬减轻神经细胞的损伤,而在24~36h后加重细胞损伤;黄芪甲苷可通过抑制细胞过度自噬诱导的细胞损伤,从而发挥对受损细胞的神经保护作用。
PC12细胞;氧糖剥夺再复糖复氧;细胞自噬;细胞损伤;黄芪甲苷;保护作用
自噬(Autophagy),即自体吞噬,是近年来逐渐被认识的细胞除坏死和凋亡外的第3种死亡方式[1],是细胞受到刺激后,通过溶酶体途径降解细胞内物质的统称。正常自噬能清除体内异常蛋白质及受损或过多的细胞器,从而维持着细胞的存活、分化、发育和稳态;而自噬过度,可诱发自噬性细胞死亡,并和凋亡、坏死交互作用,使细胞损伤加重[2-4]。缺血缺氧是自噬激活的重要诱因,有研究表明,脑缺血后自噬发挥了双刃剑的作用。在轻度缺氧等情况下,自噬能够通过降解损伤的蛋白和细胞器为合成新的蛋白提供原料和能量,从而对组织细胞发挥修复作用;若损伤过重,如重度缺氧,或组织细胞损伤时间长,自噬过度激活,则促进对组织细胞的损伤[5-7]。目前通过采用PC12细胞建立氧糖剥夺(或复糖复氧)模型,模拟脑缺血(或再灌注)后研究神经细胞自噬的变化已有一些报道[8-13]。但氧糖剥夺/复糖复氧后不同时间,PC12细胞自噬发挥了怎样的动态变化?这种改变与氧糖剥夺/复糖复氧导致的神经细胞损伤具有何种关系?对这些问题都不清楚。黄芪是治疗心脑血管疾病的常用中药,黄芪总苷(Astragalosides,AST)是黄芪中具有心脑血管药理作用的药效物质,主要含黄芪甲苷(Astragaloside IV)。已有研究表明AST及其有效成分AstragalosideⅣ具有抗脑缺血后氧化损伤[14-16]。我们前期研究也表明,AST抗缺血再灌注后神经细胞损伤的作用可能与其改善脑组织能量代谢、通过抑制JNK信号转导通路活化从而抑制线粒体凋亡途径有关[17]。但其对脑缺血再灌注后神经细胞自噬性损伤有何影响,应当以什么样的剂量可以更好地达到抗缺血性脑损伤的效果,都还不清楚。因此,本研究首先探讨了PC12细胞氧糖剥夺再复糖复氧不同时间点细胞自噬与损伤的关系,确定自噬引起细胞产生损伤的氧糖剥夺复糖复氧的可能时间点,并制作PC12细胞氧糖剥夺再复糖复氧产生自噬性损伤的细胞模型;然后再在此基础上进行黄芪甲苷抗自噬性损伤的作用研究,为其进一步地研究和临床的合理应用提供科学依据。
1 材料
1.1试剂 神经生长因子(Nerve Growth Factor, NGF)(美国sigma,批号N2513);蛋白裂解液(中国CW Biotech,批号0414K);蛋白酶抑制剂(中国CW Biotech,批号0815A);二甲基亚砜(Dimethyl Sulphoxide,DMSO)(美国MP Biomedicals,批号196055);噻唑蓝(3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide,MTT)(中国Solarbio公司,批号M8181);乳酸脱氢酶(LDH)试剂盒(中国南京建成生物有限公司,批号20140327);兔抗大鼠微管相关蛋白3(microtubule-associated protein 1 light chain 3,LC3)一抗(日本MBL,批号PM036);兔抗大鼠sequesto-some-1(SQSTM1/P62)一抗(美国Proteintech,批号18420-1-AP);小鼠抗大鼠β-actin一抗(美国Proteintech,批号60008-1-lg);荧光素四甲基异硫氰酸罗丹明(tetraethylrhodamineisothiocyanate, TRITC)标记羊抗兔二抗(中国Boster,批号: BA1090);Hochest 33258(中国Solarbio,批号COO20 10);辣根过氧化物酶(Horseradish Peroxidase,HRP)标记的羊抗兔、羊抗小鼠二抗(美国proteintech,批号SA00001-1、SA00001-2);3-Methyladenine(3-MA)(美国selleck,批号S2767)。
1.2受试药物 黄芪甲苷(批号:A0070),购自中国成都曼思特生物科技有限公司,纯度≥98%。黄芪甲苷以含0.1%DMSO-PBS溶解。
1.3细胞 PC12细胞,来源于大鼠的肾上腺嗜铬细胞瘤细胞,高分化,永生性,在美国模式菌种收集中心(American Type Culture Collection,ATCC)中的编号为CRL-1721,购自武汉大学中国典型培养物保存中心。-
2 方法
2.1PC12细胞的培养与NGF诱导 PC12细胞以含10%胎牛血清的DMEM高糖培养基于37℃、20%O2、75%N2、5%CO2及饱和湿度的培养箱中培养,2d换液1次,待细胞生长融合后传代培养。细胞以1×104/mL接种于96孔培养板,每孔0.18 mL,培养24h待细胞贴壁后再加终浓度50 μg/L的NGF诱导分化48h后[18],弃原培养液,加入无血清培养基培养24h,使细胞同步化到G0期后备用。
2.2PC12细胞氧糖剥夺复糖复氧后自噬的经时性变化及与损伤的关系
2.2.1实验分组及氧糖剥夺后复糖复氧 实验分为正常对照组、氧糖剥夺后复糖复氧模型组(缺糖缺氧2h后复糖复氧6h、12h、24h、36h)和自噬抑制剂组。细胞以DMEM高糖培养液(含10%胎牛血清和1%青链霉素)接种于96孔培养板,每孔200 μL, 置37℃、20%O2、75%N2、5%CO2及饱和湿度的培养箱中常规培养2h后进行如下处理:正常对照组换DMEM高糖培养液同上常规培养。模型组加200L的无糖Earle’s培养液,置于三气培养箱中(5% CO2,1%O2,94%N2)模拟缺糖缺氧培养2h后[19],换成DMEM高糖培养液常规培养进行复糖复氧,分别于复糖复氧后6h、12h、24h、36h终止培养进行检测。自噬抑制剂组于缺糖缺氧前30min加入终浓度25 mmol/L[20-21]的3-MA,再进行缺糖缺氧和复糖复氧培养。
2.2.2细胞生存测定和LDH漏出率测定 采用MTT法。细胞上述处理完成后,加入20 μL 5 mg/ mL的MTT继续培养4h,弃上清后加200 μL的DMSO,震荡10min后于490 nm处检测光密度值(OD)。OD值越大,则细胞存活越多,细胞损伤越少。细胞上述处理完成后,分别吸取培养液,1 000 r/min离心10min,取上清检测培养液中的LDH活性。然后将细胞用PBS洗涤3次后再用等量培养液悬浮,置-20℃、37℃反复冻融3次,再将冻融液1 000 r/min离心10min,取上清检测作为细胞冻融液的LDH活性。按LDH试剂盒说明方法测定LDH活性,并计算出细胞LDH漏出率[21]:LDH漏出率(%)=培养液LDH活性/(培养液LDH活性+细胞冻融液LDH活性)×100%。
2.2.3自噬体形态和数量测定:PC12细胞以1× 104/mL接种于6孔板中,细胞培养及处理同前,吸去上清,PBS洗1次后加入2.5%戊二醛固定液固定过夜,1%锇酸后固定2h,依次以50%、70%、90%、100%的丙酮脱水,包埋、染色后以透射电子显微镜(美国FEI,型号Tecnai G2 spirit)观察单位面积细胞内自噬体数目和形态。
2.2.4细胞LC3Ⅱ定位及相对定量检测:正常情况下LC3蛋白主要以LC3-Ⅰ形式散在分布于胞浆中,激光共聚焦显微镜检测LC3蛋白可见胞质中出现弥散状荧光;若细胞出现自噬则表现为LC3-Ⅱ的形成和在胞浆的聚集,激光共聚焦显微镜检测LC3-Ⅱ蛋白可见胞质中出现斑片状荧光体。PC12细胞以1× 104/mL接种于24孔板中,制作细胞爬片,细胞培养及处理同前,吸去上清,PBS洗一次后加4%多聚甲醛固定15min,PBS洗3次,加0.5%曲拉通10min 后,PBS洗3次,再加1%牛血清白蛋白封闭液封闭1h后,加兔抗大鼠微管相关蛋白LC3一抗(1∶500) 4℃孵育过夜,PBS洗3次,加TRITC标记的羊抗兔荧光二抗(1∶100)室温孵育30min,PBS洗3次,加Hochest33258(1∶200)10min,PBS洗3次,甘油封片后于激光共聚焦显微镜下(日本NiKon,型号A1130309)观察斑片状荧光体。Image Pro-Plug6.0图像分析软件测定细胞面积及斑片状荧光体个数,计算单位面积的斑片状荧光体个数。
2.2.5细胞LC3、p62蛋白定量检测 以western blot法测定。PC12细胞以1×104/mL接种于6孔板中,细胞培养及处理同前,吸去上清,PBS洗一次后加500 μL的PBS,用细胞刮刮下细胞,1 000 r/min离心5min,弃去上清,加入100 μL的蛋白裂解液和1 μL的蛋白酶抑制剂,冰上充分混匀后静置20min 后12 000 r/min离心10min,取上清,BCA试剂盒测定总蛋白含量。取30 μg蛋白100℃水浴10min变性后,120 v恒压电泳70min后,200 mA转膜2h, 5%脱脂牛奶封闭1h,再分别与兔抗大鼠LC3一抗(1∶500)、兔抗大鼠p62一抗(1∶500)和小鼠抗大鼠β-actin一抗(1∶1 000)溶液混合,4℃静置过夜,TBS 洗3次后TBST洗1次;然后分别加羊抗兔二抗(1∶2 000)或羊抗小鼠二抗(1∶2 000),37℃孵育1h, TBS洗3次后TBST洗1次,暗室中加ECL化学发光剂显影。Image Pro-Plug6.0图像分析软件测定目的条带的累积光密度值(IOD),以目的条带的IOD与β-actin条带的IOD的比值作为该目的蛋白的相对表达量。自噬发生后,胞浆型LC3(即LC3-I,分子量为18kD)会酶解切掉一小段多肽,转变为(自噬体)膜型LC3(即LC3-II,分子量为16 kD),通过Western-blot可以检测到两个条带的蛋白质,用LC3-II相对表达量与LC3-I相对表达量的比值代表自噬水平的高低。
2.3OGD/R自噬性损伤模型的建立及黄芪甲苷对细胞自噬的抑制作用 根据上述2.2研究结果,确定PC12细胞OGD 2h复糖复氧发生自噬性损伤的时间点为复糖复氧24~36h,故以氧糖剥夺2h复糖复氧24h制作细胞OGD/R自噬性损伤模型,同时进行药物作用研究。实验分为正常对照组、模型组和药物干预组。细胞培养及造模方法同前。药物组于缺糖缺氧前30mindMEM高糖培养液中加入黄芪甲苷0、9.81、19.63、39.25、78.5 μg/mL[22]以及阳性对照药3-MA后,加200 μL的无糖Earle's培养液,置于三气培养箱中(5%CO2,1%O2,94%N2)模拟缺糖缺氧培养2h,换成DMEM高糖培养液常规培养继续复糖复氧24h。复糖复氧24h后按2.2.4做细胞LC3Ⅱ定位及相对定量检测。以(模型组斑片体数量-药物组斑片体数量)/模型组斑片体数量×100%,计算药物对自噬的抑制率;以药物浓度为自变量,抑制率为因变量进行曲线回归分析(即以药物浓度的自然对数为自变量,抑制率为因变量),并计算药物的半数抑制浓度(IC50)和95%的可信区间。
2.4黄芪甲苷对PC12细胞OGD/R自噬性损伤的保护作用 通过MTT、LDH漏出率探讨黄芪甲苷对PC12细胞OGD/R自噬性损伤的作用。细胞照2.3处理完成后,将实验分为正常对照组、氧糖剥夺后复糖复氧模型组(缺糖缺氧2h复糖复氧24h)、黄芪甲苷(3.45 μg/mL)组及阳性对照药物3-MA组。药物处理和检测方法同前。
2.5统计学方法 实验数据用“x¯±s”表示。用SPSS 17.0进行统计分析,多组间均数比较用单因素方差分析,组间两两比较方差齐者用LSD检验,方差不齐者用DunnetT3检验。P<0.05为差异有统计学意义。
3 结果
3.1不同时间点细胞存活情况和LDH漏出率的比较 与正常组比较,缺糖缺氧2h复糖复氧6h、12h、24h、36h各模型组及抑制剂组细胞存活显著降低(均P<0.01)。复糖复氧24h、36h组细胞存活显著低于6h、12h组(均P<0.01)。与同时间点模型组比较,抑制剂组6h、12h细胞存活显著低于模型组(P<0.05或P<0.01),24h、36h细胞存活显著高于模型组(P<0.01)(图1)。
与正常对照组比较,缺糖缺氧2h复糖复氧6h、12h、24h、36h后细胞LDH漏出率增加(P<0.05或P<0.01),以24h最高,且显著高于复糖复氧6h、12h(P<0.01)。与同时间点模型组比较,抑制剂组12hLDH漏出率显著升高(P<0.01),24h 和36h LDH漏出率显著降低(均P<0.01)(图1)。

图1 各组复糖复氧后不同时间细胞存活率和乳酸脱氢酶(LDH)漏出率比较(±s,n=3)
3.2不同时间点细胞自噬体的形态和数量的比较以及LC3Ⅱ定位和相对定量检测 正常对照组未见明显自噬体形成。缺糖缺氧2h复糖复氧6h后胞浆内可见少量完整自噬体,呈双层膜结构包裹细胞内容物;复糖复氧12h自噬体数目较6h组明显增多,24h达高峰,且可见少量自噬体与溶酶体融合形成自噬溶酶体现象,部分自噬体内容物被降解,形成单层膜结构;复糖复氧36h自噬体体积增大,降解加重,数量减少(图2)。与正常组比较,复糖复氧6、 12h、24h、36h自噬体数目均显著增多(P<0.05或P<0.01)(图3)。正常对照组细胞胞浆内可见均匀分布的红色荧光点,无斑片状荧光体形成。OGD2h复糖复氧6h、12h、24h、36h后,胞浆内可见斑片状荧光体聚集(图4)。与正常组比较,模型组复糖复氧6h、12h、24h、36h细胞内斑片状荧光体数目显著增多(均P<0.01),其中以24h组斑片状荧光体数量最多。与同时刻模型组比较,抑制剂组斑片状荧光体数目均显著减少(P<0.05或P<0.01)(图3)。 3.3 不同时间点LC3-Ⅱ/LC3-Ⅰ及p62蛋白表达的比较 与正常对照组比较,模型组和抑制剂组复糖复氧6h、12h、24h、36h细胞LC3-Ⅱ/LC3-Ⅰ比值显著增加(均P<0.01),24h达到高峰,然后逐渐下降。与同时刻模型组比较,抑制剂组复糖复氧6h、12h、24h、36h细胞LC3-Ⅱ/LC3-Ⅰ比值均显著降低(P<0.05或P<0.01)(图5)。
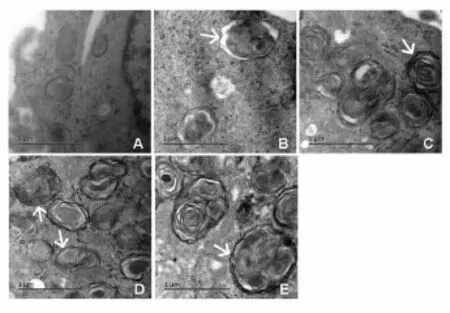
图2 各组细胞自噬体的超微形态(×30 000,Bar=1 μm)
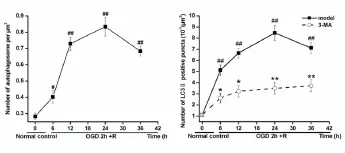
图3 各组细胞自噬体数量及LC3-Ⅱ相对定量的比较(±s,n=3)
与正常对照组比较,模型组p62蛋白表达均显著下降(均P <0.01),36h最低。与同时刻模型组比较,抑制剂组6h、12h、24h及36h细胞p62蛋白表达均显著升高(P<0.05或P<0.01)(图5)。
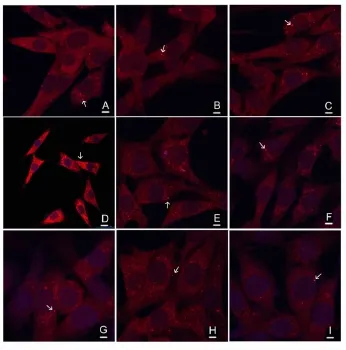
图4 各组细胞LC3Ⅱ定位(×600,Bar=20 μm)
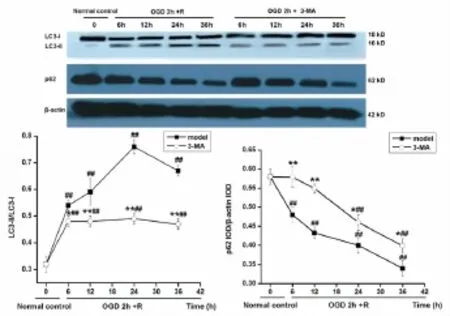
图5 各组LC3-Ⅱ/LC3-Ⅰ及P62蛋白表达比较(±s,n=3)
3.4黄芪甲苷对OGD/R细胞自噬的抑制作用 由图6可知,PC12细胞OGD 2h复糖复氧24h后,无药物干预时,对自噬的抑制率为0。而自噬抑制剂3-MA和不同浓度黄芪甲苷对自噬有显著的抑制作用,与模型组比较差异有统计学意义(P <0.01);且黄芪甲苷对自噬的抑制作用随着药物剂量的增加而逐步增强,经曲线回归分析得出黄芪甲苷抑制自噬的IC50为(27.22±0.614)μg/mL,95%可信区间为[25.96,29.03]μg/mL。
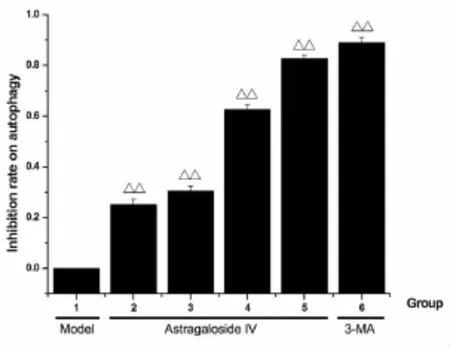
图6 不同剂量的黄芪甲苷对自噬的抑制率(±s,n=3)
3.5IC50的黄芪甲苷对氧糖剥夺复糖复氧后PC12细胞存活和LDH漏出率影响 MTT细胞存活实验结果表明,与正常对照组比较,模型组细胞存活显著减少,LDH漏出率显著增加(P<0.01)。与模型组比较,黄芪甲苷组、3-MA组细胞存活均显著增加LDH而漏出率显著降低(P<0.01)(图7)。

图7 各组细胞存活、LDH漏出率数量的比较(±s,n=3)
4 讨论
自噬过程是指从粗面内质网的无核糖体附着区脱落的双层膜或高尔基体膜包裹部分胞质和细胞内需降解的细胞器、蛋白质等成分形成自噬体(Autophagosome),并与溶酶体融合形成自噬溶酶体,降解其所包裹的内容物,以实现细胞本身的代谢需要和某些细胞器更新的过程。微管相关蛋白3(Microtubule-associated Protein 1 Light Chain 3,LC3)是Atg8基因的同源物,自噬产生过程中LC3在半胱氨酸蛋白酶Atg4的作用下脱去羟基端变成LC3-Ⅰ,随后LC3-Ⅰ被Atg7激活并转位到Atg3,在Atg3的作用下与磷脂酰乙醇胺连接并酯化形成LC3-II,定位于自噬体的内膜和外膜,参与自噬体的形成;LC3-Ⅱ始终稳定地保留在自噬体膜上直到与溶酶体融合,因此被用来作为自噬体的标记,LC3-Ⅱ的水平和LC3-II/LC3-I比值的大小在某种程度上反映了自噬体的数量和细胞自噬水平的高低[23-25]。p62是一种多功能泛素结合蛋白,参与了细胞信号转导和功能的调节,其中LRS结构域由11个氨基酸(Ser334-Ser344)组成,供LC3-II识别,为自噬形成过程中LC3-II蛋白的底物,在自噬小体形成过程中作为连接LC3和泛素化蛋白之间的桥梁,与定位于自噬小体内膜上的LC3-II蛋白形成复合物,并通过泛素信号途径将聚合的蛋白、受损的细胞器、错误折叠或聚合的蛋白以及某些长寿分子、入侵的细菌作为受体转运到自噬小体,一同在自噬溶酶体(Autolososome)中降解。因此p62参与了自噬形成过程的选择性自噬,可结合泛素,也可与LC3结合,从而靶向自噬体并促进泛素化蛋白的清除和自身的特异性降解,是反映自噬活性的标记蛋白之一,与LC3-II共同参与自噬活性的判断;自噬过程受损时通常伴有细胞内P62的累积[26-28]。
本研究结果显示,PC12细胞在缺糖缺氧2h复糖复氧6h、12h、24h、36h后,细胞存活率显著减少、LDH漏出率显著增加,表明产生了细胞损伤。在复糖复氧后6~12h,以自噬抑制剂干预后,细胞存活率降低,LDH漏出率升高,细胞损伤加重;而在复糖复氧后24~36h,自噬抑制剂可使细胞存活率增加,LDH漏出率减少,细胞损伤减轻。表明自噬在PC12细胞氧糖剥夺后再复糖复氧中产生了双向作用,即在复糖复氧后6~12h可能对细胞起保护作用,在复糖复氧后24~36h对细胞具有损伤作用。透射电镜结果显示,缺糖缺氧2h复糖复氧6h后即可见自噬体形成,复糖复氧12~24h可见大量自噬体聚集,复糖复氧36h还可见自噬溶酶体,并伴随自噬体降解自身和细胞内容物的现象,自噬体数量有所减少。激光共聚焦显微镜检测LC3蛋白定位和western blot检测LC3蛋白表达,结果也显示,在复糖复氧6h,即出现LC3向自噬体膜的转位、LC3-Ⅱ/ LC3-Ⅰ比值增加,表明自噬被诱导;复糖复氧12~ 24h,LC3向自噬体膜的转位增多,LC3-Ⅱ/LC3-Ⅰ比值进一步增加,表明自噬程度逐渐增强;至复糖复氧36h,LC3-Ⅱ向自噬体膜的转位及LC3-Ⅱ/LC3-Ⅰ有所降低,自噬体数量减少,但仍高于正常对照。可能是在复糖复氧36h后,由于自噬体活性过度增强,使细胞内LC3-Ⅱ蛋白降解,从而使转位的LC3-Ⅱ减少及LC3-Ⅱ/LC3-Ⅰ比值降低。用自噬抑制剂3-MA干预后,复糖复氧各时间点LC3-Ⅱ转位及LC3-Ⅱ/LC3-Ⅰ均有所减少,自噬减轻。表明自噬抑制剂可抑制复糖复氧诱导的自噬激活。p62蛋白表达结果也显示,在复糖复氧6h、12h、24h及36h p62蛋白表达逐渐降低;3-MA预处理后,可使各时间点p62蛋白表达增强。表明随着复糖复氧时间的延长,自噬活性逐渐增强,导致p62蛋白逐渐降解。
由以上研究结果可知,在缺糖缺氧2h复糖复氧6~12h自噬对细胞模拟的缺血再灌注损伤可能起保护作用,而在复糖复氧24~36h后可能起损伤作用。故我们在后继研究中选用缺糖缺氧2h复糖复氧24h制作PC12细胞OGD/R自噬性损伤模型,结果表明,PC12细胞在OGD 2h复糖复氧24h后,细胞存活减少,LDH漏出率和自噬体数量增多。不同剂量的黄芪甲苷可使自噬体数量减少,且黄芪甲苷对自噬的抑制率随着剂量的加大而增加,表明黄芪甲苷对OGD/R诱导的细胞自噬具有抑制作用。黄芪甲苷的IC50为(3.45±0.573)μg/m,在该剂量下,黄芪甲苷可使OGD 2h复糖复氧24h后的PC12细胞存活增加,LDH漏出率减少。
综上所述,缺糖缺氧2h再复糖复氧24h后,细胞出现过度自噬和损伤,提示细胞过度自噬参与了对细胞的损伤,黄芪甲苷可通过对细胞自噬性损伤的拮抗,从而发挥对受损神经元的保护作用。具体机制有待进一步研究。
[1]QinhD,Tan WG,Zhang Z,et al.15d-Prostaglandin J2 Protects Cortical Neurons Against Oxygen-Glucosedeprivation/Reoxygenation Injury:Involvement of Inhibiting.Autophagy Through Upregulation of Bcl-2[J].Cell Mol Neurobiol,2015,35:303-312.
[2]Uchiyama Y,Shibata M,Koike M,et al.Autophagy-physiology and pathophysiology[J].Histochem Cell Biol,2008,129(4):407-420.
[3]Kroemer G,Mariño G,Levine B.Autophagy and the integrated stress response[J].Mol Cell,2010,40(2):280-293.
[4]Li L,Zhang Q,Tan J,et al.Autophagy andhippocampal neuronal injury[J].Sleep Breath,2014,18(2):243-249.
[5]Li L,Tan J,Miao Y,et al.ROS and Autophagy:Interactions and Molecular Regulatory Mechanisms[J].Cell Mol Neurobiol,2015,35(5):615-621.
[6]Huang XP,Dingh,Lu JD,et al.Autophagy in cerebral ischemia and the intervention effects of traditional Chinese medicine[J].J Integr Med,2015,13(5):289-296.
[7]Wei K,Wang P,Miao CY.Adouble-edged sword with therapeutic potential:an updated role of autophagy in ischemic cerebral injury[J]. CNS Neuroscience&Therapeutics,2012,18:879-886.
[8]Zhang XY,Zhang TT,SongdD,et al.Endoplasmic reticulum chaperone GRP78 is involved in autophagy activation induced by ischemic preconditioning in neural cells[J].Mol Brain,2015,26,8:20.
[9]Zuo W,Zhang S,Xia CY,et al.Mitochondria autophagy is induced afterhypoxic/ischemic stress in adrp1dependent manner:the role of inhibition ofdrp1 in ischemic braindamage[J].Neuropharmacology, 2014,86:103-115.
[10]Mo Z,Fang Y,He Y,et al.Change of Beclin-1dependent on ATP, [Ca(2+)](i)and MMP in PC12 cells following oxygen-glucosedeprivation-reoxygenation injury[J].Cell Biol Int,2012,36(11): 1043-1048.
[11]Mo ZT,Fang YQ,He YP,et al.β-Asarone protects PC12 cells against OGD/R-induced injury via attenuating Beclin-1-dependent autophagy[J],Acta Pharmacologica Sinica,2012,33:737-742.
[12]Derong Cui,Li Wang,Aihua Qi,et al.Propofol Prevents Autophagic Celldeath following Oxygen and Glucosedeprivation in PC12 Cells and Cerebral Ischemia-Reperfusion Injury in Rats[J].PloS One, 2012,7(4):e35324.
[13]Chiu BY,Chang CP,Lin JW,et al.Beneficial Effect of Astragalosides on Stroke Condition Using PC12 Cells under Oxygen Glucosedeprivation and Reperfusion[J].Cell Mol Neurobiol,2014,34:825-837.
[14]Yin YY,Li WP,GonghL,et al.Protective effect of astragaloside on focal cerebral ischemia/reperfusion injury in rats[J].Am J Chin Med,2010,38:517-527.
[15]Qu YZ,Li M,Zhao YL,et al.Astragaloside IV attenuates cerebral ischemia-reperfusion induced increase in permeability of the bloodbrain barrier in rats[J].Eur J Pharmacol,2009,606:137-141.
[16]Li WZ,Li WP,Zhang W,et al.Protective effect of extract of astragalus on learning and memory impairments and neurons apoptosis induced by glucocorticoids in 12-month male mice[J].Anat Rec (Hoboken),2011,10.1002/ar.21386.
[17]Huang XP,Tanh,Chen BY,et al.Astragalus extract alleviates nerve injury after cerebral ischemia by improving energy metabolism and inhibiting apoptosis[J].Biol Pharm Bull,2012,35(4):449-454.
[18]He Li,Alexander Ruvantha Pinto,Duan WZ,et al.Telomerasedownregulationdoes not mediate PC12 pheochromocytoma celldifferentiation induced by NGF,but requires MAP kinase signaling[J].Journal of Neurochemistry,2005,95,891-901.
[19]Zhang WD,Smith C,Shapiro A,et al.Increased expression of bioactive chemokines inhuman cerebromicrovascular endothelial cells and astrocytes inhuman cerebromicrovascular endothelial cells and astrocytes subjected to simulated ischemia in vitro[J].Neuroimmunology,1999,101:148-160.
[20]LihY,AJ,FengdX,et al.Evaluation of the protective potential of Brain Microvascular Endothelial cell Autophagy on Blood-Brain Barrier Integrityduring Experimental Cerebral Ischemia-Reperfusion Injury[J].Transl.Stroke Res,2014,5:618-626.
[21]Sheng R,Liu XQ,Zhang LS,et al.Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning[J].Autophagy,2012,8 (3):310-325.
[22]刘晓丹,邓常清.黄芪甲苷和三七总皂苷中主要有效成分抗PC12细胞氧化损伤的配伍研究[J].湖南中医药大学学报, 2012,32(1):8-12.
[23]Xu F,Gu JH,Qin ZH.Neuronal autophagy in cerebral ischemia[J]. Neurosci Bull,2012,28(5):658-666.
[24]Kimura S,Fujita N,Noda T,et al.Monitoring autophagy in mammalian cultured cells through thedynamics of LC3[J].Methods Enzymol,2009,452:1-12
[25]Barth S,Glickd,Macleod KF.Autophagy:assays and artifacts[J].J Pathol,2010,221(2):117-124.
[26]Bjorkoy G,Lamark T,Pankiv S,et al.Monitoring autophagicdegradation of p62/SQSTM1[J].Methods Enzymol,2009,452:181-197
[27]BenYounès A,Tajeddine N,Tailler M,et al.A fluorescencemicroscopic and cytofluorometric system for monitoring the turnover of the autophagic substrate p62/SQSTM1[J].Autophagy,2011,7(8):883-891.
[28]Johansen T,Lamark T.Selective autophagy mediated by autophagic adapter proteins[J].Autophagy,2011,7(3):279-296.
(2016-03-24收稿 责任编辑:洪志强)
Relationship Between Autophagy anddamage in the Oxygen Glucosedeprivation Model of PC12 Cells and the Protective Effect of Astragaloside IV
Huang Xiaoping,Li Jingxian,Yang Xiaoqian,Dinghuang,Tang Biao,Liu Xiaodan,Deng Changqing
(Molecular Pathology Laboratory,Key Laboratory ofhunan Province for Prevention and Treatment of Integrated Traditional Chinese and Western Medicine on Cardio-Cerebraldiseases,Key Laboratory ofhunan Universities for Cell Biology and Molecular Techniques,Hunan University of Chinese Medicine,Changsha 410208,Hunan)
Objective:To explore the changes of autophagy and injury indifferent time periods after PC12 cell were induced by oxygen-glucosedeprivation/reoxygenation(OGD/R),and the intervention effects of Astragaloside IV.Methods:Firstly,the changes of autophagy atdifferent time periods and the relationship with celldamage after PC12 cell oxygen-glucosedeprivation/reoxygenation(OGD/R)was probed todetermine the point that autophagy contributed to cell injury,then the the injury model of PC12 OGD/R was established to investigate the effect of Astragaloside IV against autophagy injury.Results:After reoxygenation for 6~36h,cell survival ratedecreased,the leakage rate of LDH increased.Pretreated with 3-mA,cell survival ratedecreased significantly,LDH leakage rate increased remarkably after reoxygenation for 6~12h,while being opposite after reoxygenation 24~36h.Confocal microscopy and western-blot showed that the autophagosome was formed after reoxygenation for 6h and LC3-Ⅱ,LC3-Ⅱ/LC3-Ⅰwas increased,getting to the maximum at 24h after reoxygenation.The expression of p62 proteindecreased gradually along with time.Astragaloside IV could remarkably inhibit cell autophagy followed by PC12 cell OGD/R 24h and presenteddosedependence.Conclusion:Autophagy may reduce thedamage of nerve cells at 6~12h after reoxygenation,however,excessive autophagy aggravates thedamage after reoxygenation for 24~36h;Astragaloside IV exerts the protective effect from the injured cells through inhibiting the celldamage induced by excessive autophagy.
PC12 cell;Oxygen glucosedeprivation/reoxygenation;Autophagy;Cell injury;Astragaloside IV;Protective effect
R965
A
10.3969/j.issn.1673-7202.2016.04.008
国家自然科学基金项目(编号:81473581;81573875);湖南省高校创新平台开放基金资助项目(编号:11K050;14K068);湖南省科技厅一般项目(编号:2014SK3001);湖南省中医药管理局重点项目(编号:201301;201508);中医内科重大疾病防治及成果转化省部共建教育部重点实验室开放课题(编号:ZYNK201405);“中西医结合防治心脑血管疾病的相关基础研究”湖南省高校科技创新团队;“中医药防治心脑血管疾病基础研究”湖南省自然科学创新群体基金
黄小平(1974.02—),女,硕士,副教授,硕士研究生导师,研究方向:心脑血管疾病防治的应用基础研究,Tel:(0731)88458258,E-mail:569229858@qq.com
邓常清(1963.02—),男,医学博士,教授,研究方向:防治心脑血管疾病中药及复方的药效物质和配伍原理研究,E-mail:dchangq @sohu.com
