12-对甲基苯酰氧基-14-脱氧穿心莲内酯合成工艺改进*
2016-09-01李晶,杨超,张雷
李 晶,杨 超,张 雷
(1 华南理工大学生物科学与工程学院,广东 广州 510006;2 广州白云山中一药业有限公司,广东 广州 510530)
12-对甲基苯酰氧基-14-脱氧穿心莲内酯合成工艺改进*
李晶1,杨超2,张雷1
(1 华南理工大学生物科学与工程学院,广东广州510006;2 广州白云山中一药业有限公司,广东广州510530)
为了较高收率地获得12-对甲基苯酰氧基-14-脱氧穿心莲内酯,对其合成工艺中的酯化反应进行优化研究。发现反应时间、原料投料比、溶剂用量、缩合剂用量分别为22 h,1:1.1,1:25,1:1.2时为佳,且使用缩合剂DIC时的反应收率最高,使用缩合剂EDAC时副产物最少。此外,对起始原料穿心莲内酯进行重结晶纯化有利于减少副产物。经上述反应优化后,总收率可达15.3%。中间体及目标产物的结构均经1H-NMR、13C-NMR、IR和MS确证。
穿心莲二萜内酯;合成;工艺改进
穿心莲为爵床科穿心莲属植物穿心莲的干燥地上部分,是我国传统中药之一,具有清热解毒、抗炎抗病毒、镇痛等功效[1]。其主要活性成分是以穿心莲内酯为代表的二萜内酯类化合物。研究发现穿心莲内酯可以共价键与p50亚单位结合,抑制转录因子NF-кB活化,阻断NF-кB与DNA的结合[2-3],从而抑制巨噬细胞中多种炎症因子基因的表达,如细胞间黏附分子-1[4]、NO、iNOS[2]、COX-2[5-6]、TNF-α[7]、INF-γ、IL-6、IL-2等[3]。因此,穿心莲内酯作为干预炎症反应的重要天然产物,其结构修饰与改造吸引了众多药学工作者的目光。
我们的前期研究发现12-对甲基苯酰氧基-14-脱氧穿心莲内酯对LPS诱导的巨噬细胞过度表达TNF-α、IL-6等炎症介质具有很好的抑制活性[8],可以作为穿心莲内酯的先导优化物进行深入开发。但其合成方法[9]报道较少。我们按文献方法合成12-对甲基苯酰氧基-14-脱氧穿心莲内酯,即以穿心莲内酯(1)为起始原料,对甲苯磺酸吡啶盐(PPTs)催化下,与2,2-二甲氧基丙烷反应制得3,19-异亚丙基穿心莲内酯(2);2经重铬酸吡啶嗡盐(PDC)作用发生重排反应生成3,19-异亚丙基-12-羟基-14-脱氧穿心莲内酯(3);3与对甲基苯甲酸在缩合剂二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP)催化下进行酯化反应得到3,19-异亚丙基-12-对甲基苯甲酰基-14-脱氧穿心莲内酯(4);4在酸性条件下水解脱去缩酮保即得目标产物12-对甲基苯酰氧基-14-脱氧穿心莲内酯(5)(Scheme 1)。该法总收率较低,为2.3%。尤其是酯化反应一步,反应时间长,收率低,副产物非常多且不易纯化。
本文重点对酯化反应这一步进行研究,采用其他缩合剂DIC、EDAC替代DCC,比较不同缩合剂对该步反应收率的影响,此外,对该反应的反应时间、酸醇投料比、缩合剂用量等也进行了考察。改进后的新工艺具有操作步骤简单、反应条件温和,且总收率有较大的提高,能达到15.3%。另外,该方法对穿心莲内酯3-OH和19-OH的缩合反应也具有一定的指导意义。
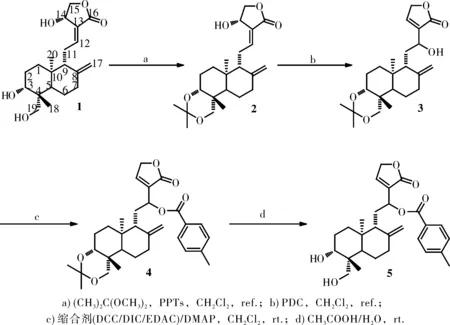
图1 12-对甲基苯酰氧基-14-脱氧穿心莲内酯的合成
1 实验部分
1.1仪器与试剂
1100型高效液相色谱仪,检测波长为224 nm,流动相为乙腈和水(20,80~90,10);Tensor 27傅里叶变换红外光谱仪;Bruker Avance III 400型核磁共振谱仪;质谱仪;HH-4数显恒温油浴锅;85-1B磁力搅拌器;R-200旋转蒸发仪。
穿心莲内酯,安徽拓峰生物制品厂产品;2,2-二甲氧基丙烷、4-二甲氨基吡啶(DMAP)、二环己基碳二亚胺(DCC)、二异丙基碳化二亚胺(DIC)、1-乙基-3(3-二甲基胺丙基)碳化二亚胺(EDAC),均为百灵威科技有限公司产品;对甲基苯甲酸为化学纯,其他试剂均为分析纯。硅胶100~200目,青岛谱科分离材料有限公司产品。
1.2合成实验
1.2.13,19-异亚丙基穿心莲内酯(2)
将穿心莲内酯(1,10.0 g,28.7 mmol)溶解在二氯甲烷150 mL中,加入2,2-二甲氧基丙烷(13.3 mL,108.7 mmol)与催化量对甲苯磺酸吡啶盐(PPTs)。加热回流30 min。反应结束后冷至室温。滴加三乙胺调pH至8左右,加入200 mL二氯甲烷稀释反应液,以饱和食盐水(200 mL×3)洗,无水硫酸钠干燥,过滤浓缩,有固体析出,抽滤,滤饼干燥得粗产品,二氯甲烷重结晶得白色固体(9.6 g,85.5%)。1H-NMR (CDCl3, 400 MHz) δ 6.94(1H, td,J=12.0 Hz, H-12), 5.03(1H, t,J=12.0 Hz, H-14), 4.90(1H, s, H-17a), 4.62(1H, s, H-17b), 4.44(1H, dd,J=4.0 Hz, 8.0 Hz, H-15a), 4.25(1H, dd,J=4.0 Hz, 2.0 Hz, H-15b), 3.96(1H, d,J=12.0 Hz, H-19a), 3.49(1H, dd,J=4.0 Hz, 4.0 Hz, H-3), 3.18(1H, m, H-19b), 2.58(3H, m, H-1, 11a), 2.41(1H, m, H-11b), 1.98(2H, m, H-7), 1.76(4H, m, H-2, 6), 1.29(11H, m, H-5, 9, 18, -C(CH3)2), 0.95(3H, s, H-20);13C-NMR (CDCl3, 100 MHz) δ 170.20, 149.15, 147.06, 128.03, 108.98, 99.23, 76.23, 74.44, 66.19, 63.96, 56.08, 52.25, 38.48, 37.96, 37.70, 34.59, 27.10, 26.18, 25.37, 25.07, 25.00, 23.25, 16.25; IR(KBr, cm-1) ν 3479, 3077, 2986, 2937, 2864, 1747, 1685, 1641, 1460, 1379, 1223, 1066, 1020, 988, 901, 859; EI-MS: 517.5[M+(CH3)2C(OCH3)2+Na]+。
1.2.23,19-异亚丙基-12-羟基-14-脱氧穿心莲内酯(3)
将2(8.0 g,20.6 mmol)溶解在二氯甲烷150 mL中,逐渐加入重铬酸吡啶嗡盐(PDC)(10.1 g,26.7 mmol),约2 h加完,室温搅拌30 min, TLC监测。反应完毕,蒸干溶剂,再用乙酸乙酯(50 mL×3)溶解,过滤,硅胶柱层析分离(石油醚/乙酸乙酯=3:2),得淡棕色油状物(3.5 g,43.6%)。1H-NMR (CDCl3, 400 MHz) δ 7.27(1H, overlapped single, H-14), 4.89(1H, s, H-17a), 4.80(2H, s, H-15), 4.72(1H, s, H-17b), 4.51(1H, t,J=12.0 Hz, H-12), 3.92(1H, d,J=12.0 Hz, H-19a), 3.43(1H, dd,J=4.0 Hz, 4.0 Hz, H-3), 3.13(1H, d,J=12.0 Hz, H-19b), 2.86(1H, s, H-OH), 2.38(1H, m, H-11a), 1.92(4H, m, H-1, 7a, 11b), 1.72(4H, m, H-2, 6), 1.59(1H, m, H-7b), 1.25(11H, m, H-5, 9, 18, -C(CH3)2), 0.87(3H, s, H-20);13C-NMR (CDCl3, 100 MHz) δ 173.00, 148.56, 145.38, 136.20, 107.88, 99.03, 76.43, 70.47, 67.40, 63.82, 53.02, 52.42, 38.68, 38.06, 37.84, 34.28, 30.53, 27.24, 26.06, 25.28, 25.02, 23.44, 16.27; IR(KBr, cm-1) ν 3448, 3081, 2986, 2939, 2894, 1749, 1644, 1449, 1376, 1228, 1093, 833; EI-MS: 413.1[M+Na]+。
1.2.33,19-异亚丙基-12-对甲基苯甲酰基-14-脱氧穿心莲内酯(4)
方法一:将3 (1.0 g,2.60 mmol)溶解在二氯甲烷25 mL中,加入对甲基苯甲酸(0.39 g,2.86 mmol)与催化剂量的DMAP,室温搅拌30 min。在冰浴条件下,逐渐加入DCC (0.65 g,3.12 mmol),室温搅拌24 h, TLC监测。反应完毕,过滤,滤液蒸去溶剂,残余物用25 mL乙酸乙酯溶解,置于冰箱过夜,过滤,硅胶柱层析分离(石油醚/乙酸乙酯=4:1),得白色固体(0.12g,9.6%)。1H-NMR (CDCl3, 400 MHz) δ 7.92(2H, d, Ar-H), 7.39(1H, s, H-14), 7.24(2H, d, Ar-H), 5.89(1H, m, H-12), 4.93(1H, s, H-17a), 4.84(3H, m, H-15, 17b), 3.96(1H, d,J=12.0 Hz, H-19a), 3.46(1H, m, H-3), 3.16(1H, d,J=12.0 Hz, H-19b), 2.36(5H, m, H-11, Ar-CH3), 1.88(8H, m, H-1, 2, 6, 7), 1.28(11H, m, H-5, 9, 18, -C(CH3)2), 0.90(3H, s, H-20);13C-NMR (CDCl3, 100 MHz) δ 171.56, 165.71, 148.15, 147.15, 144.07, 132.99, 129.84, 129.21, 127.12, 108.25, 99.03, 76.67, 70.15, 69.12, 63.86, 52.62, 52.54, 38.73, 38.01, 37.87, 34.34, 27.57, 27.46, 26.13, 25.39, 25.18, 23.45, 21.75, 16.25; IR(KBr, cm-1) ν 3094, 3002, 2935, 2851, 1754, 1717, 1644, 1613, 1578, 1510, 1440, 1298, 1264, 1043, 752; EI-MS: 531.2[M+Na]+。
方法二:将3 (1.0 g,2.60 mmol)溶解在二氯甲烷25 mL中,加入对甲基苯甲酸(0.39 g,2.86 mmol)与催化剂量的DMAP,室温搅拌30 min。在冰浴条件下,逐渐加入DIC (0.40 g,3.12 mmol),室温搅拌22 h, TLC监测。后处理同方法一,得白色固体(0.61 g,48.2%)。
方法三:将3 (1.0 g,2.60 mmol)溶解在二氯甲烷25 mL中,加入对甲基苯甲酸(0.39 g,2.86 mmol)与催化剂量的DMAP,室温搅拌30 min。在冰浴条件下,逐渐加入EDAC (0.61 g,3.12 mmol),室温搅拌22 h, TLC监测。后处理同方法一,得白色固体(0.56 g,44.3%)。
1.2.412-对甲基苯酰氧基-14-脱氧穿心莲内酯(5)
将4 (0.2 g,0.40 mmol)溶解在由3 mL乙酸和1.5 mL水组成的混合溶剂中,室温搅拌3 h。反应完毕,反应液用乙酸乙酯100 mL溶解,有机相分别以饱和碳酸氢钠溶液、饱和食盐水洗至pH为8左右,无水硫酸钠干燥,过滤,硅胶柱层析分离(石油醚/乙酸乙酯=1:1),得白色固体(0.16 g,85%)。1H-NMR (CDCl3, 400 MHz) δ 7.91(2H, d, Ar-H), 7.39(1H, s, H-14), 7.23(2H, d, Ar-H), 5.87(1H, dd,J=4.0 Hz, 4.0 Hz, H-12), 4.91(1H, s, H-17a), 4.83(3H, s, H-15, 17b), 4.15(1H, d,J=8.0 Hz, H-19a), 3.44(1H, m, H-3), 3.28(1H, d,J=12.0 Hz, H-19b), 2.35(5H, m, H-11, Ar-CH3), 2.03(1H, m, H-7a), 1.83(6H, m, H-1, 2, 6), 1.55(1H, d,J=12.0 Hz, H-7b), 1.20(5H, m, H-5, 9, 18), 0.64(3H, s, H-20);13C-NMR (CDCl3, 100 MHz) δ 171.62, 165.76, 148.48, 146.77, 144.13, 132.77, 129.82, 129.22, 127.04, 108.10, 80.50, 70.19, 69.05, 64.15, 55.28, 52.54, 42.89, 39.17, 38.09, 36.66, 28.18, 27.28, 23.93, 22.70, 21.74, 15.24; IR(KBr, cm-1) ν 3396, 3083, 2938, 2870, 1757, 1715, 1646, 1612, 1577, 1509, 1447, 1270, 1179, 1036, 838, 754; EI-MS: 491.1[M+Na]+。
2 结果与讨论
(1)本实验中的穿心莲内酯原料纯度为84%,但TLC仍发现少量的杂质,加之穿心莲内酯本身结构不稳定,化学性质活泼,这可能导致在2的合成中出现很多杂质,且很难分离,降低了目标产物的收率。因此,本文采用乙醇重结晶的方法[10]对1的纯化工艺进行了探讨,使用反相高效液相色谱法检测原料纯度。实验发现,以物料比(m(1):V(乙醇))为1:19的收率最高,能达到92%。
(2)在3的合成中,由于反应历程复杂,副产物较多,延长反应时间有利于提高收率,但通过实验研究发现,在加热回流条件下延长反应时间会增加副产物的种类和含量。因此,本文采用室温条件下延长反应时间的方法,反应时间以12 h为宜。
(3)采用文献方法[10]合成4时,由于缩合剂DCC在反应过程中与3生成的中间产物比较稳定,较难进一步转化为我们需要的酯化产物,且DCU反应后不易完全除去,致使产率较低且不易纯化。本文采用DCC、DIC、EDAC三种不同缩合剂进行对比,同时对合成反应的缩合剂的用量、醇酸投料比、溶剂用量和反应时间等因素进行了考察。实验结果显示:反应时间、醇酸投料比、溶剂用量、缩合剂用量分别为22 h,1:1.1,1:25,1:1.2时为宜。此外,我们还发现使用缩合剂DIC时,目标产物的收率最高,但其反应过程中的副产物较多;相比之下,使用EDAC时收率较DIC要低,但是其副产物少,且可以完全除去,便于分离纯化。
[1]Swapan P, Sukdeb B, Basudeb A, et al. Andropanolide and Isoandrographolide, Minor Diterpenoids from Andrographis paniculata: Structure and X-ray Crystallographic Analysis[J]. J. Nat. Prod., 2006, 69: 403-405.
[2]Chiou W F, Chen C F, Lin J J. Mechanisms of suppression of inducible nitric oxide synthase (iNOS) expression in RAW 264.7 cells by andrographolide[J]. Br. J. Pharmacol., 2000, 129: 1553-1560.
[3]Iruretagoyena M I, Tobar J A, Gonzales P A, et al. Andrographolide interferes with T cell activation and reduces experimental autoimmune encephalomyelitis in the mouse[J]. J. Pharmacol. Exp. Ther., 2005, 312: 366-372.
[4]Habtemariam S. Andrographolide inhibits the tumour necrosis factor-alpha-induced upregulation of ICAM-1 expression and endothelial-monocyte adhesion[J]. Phytother. Res., 1998, 12: 37-40.
[5]Hidalgo M A, Romero A, Figueroa J, et al. Andrographolide interferes with binding of nuclear factor-kappaB to DNA in HL-60-derived neutrophilic cells[J]. Br. J. Pharmacol., 2005, 144: 680-686.
[6]Wang T, Liu B, Zhang W, et al. Andrographolide reduces inflammation-mediated dopaminergic neurodegeneration in mesencephalic neuronglia cultures by inhibiting microglial activation[J]. J. Pharmacol. Exp. Ther., 2004, 308: 975-983.
[7]Oin L H, Kong L, Shi G J, et al. Andrographolide inhibits the production of TNF-alpha and interleukin-12 in lipopolysaccharide-stimulated macrophages: role of mitogen-activated protein kinases[J]. Biol. Pharm. Bull., 2006, 29: 220-224.
[8]徐浩, 王新扬, 黄文龙, 等. N-乙酰基-12-氨基亚甲基-14-脱氧穿心莲内酯衍生物的合成及其抗肿瘤活性[J]. 中国药科大学学报, 2005, 36(6):496-499.
[9]Li J, Huang W L, Zhang H B, et al. Synthesis of andrographolide derivatives and their TNF-a and IL-6 expression inhibitory activities[J]. Bioorg. Med. Chem. Lett., 2007, 17: 6891-6894.
[10]郭洁芬, 陈峻, 王英. 穿心莲内酯醇提取工艺[J]. 中国民族民间医药, 2009(7): 13-14.
Process Improvement on the Synthesis of 12-Methyl Benzoyloxy-14-deoxy Andrographolide*
LI Jing1, YANG Chao2, ZHANG Lei1
(1 School of Bioscience and Bioengineering, South China University of Technology, Guangdong Guangzhou 510006;2GuangzhouBaiyunshanZhongyiPharmaceuticalCo.,Ltd.,GuangdongGuanzhou510530,China)
To synthesize 12-methyl benzoyloxy-14-deoxy andrographolide in good yield, the esterification in the synthetic process was optimized. It was found that the optimum reaction time, reactant ratio, reactant/solvent ratio, and the reactant/condensation agent ratio was 22 h, 1:1.1, 1:25, 1:1.2, respectively, with the highest yield when the condensation agent was DIC, and the fewest by-product when the condensation agent was EDAC. In addition, the recrystallization of andrographolide was benefit to reduce by-product. The total yield could be over 15.3% after the above optimization. The structure of intermediates and target product were identified by IR, NMR and MS.
andrographis diterpene lactone; synthesis; process improvement
广州市科学研究专项项目(No.2014J4100015)。
李晶(1980-),女,副教授,主要从事药物设计与合成研究。
R914.5
A
1001-9677(2016)01-0059-03
