结合核磁共振技术与液质联用技术的代谢组学数据采集、处理和分析
2016-08-30李兴余玲玲胡凯锋中国科学院昆明植物研究所植物化学与西部植物资源持续利用国家重点实验室昆明650201中国科学院大学北京100049云南大学化学科学与工程学院昆明650091
李兴,余玲玲,胡凯锋(1、中国科学院昆明植物研究所植物化学与西部植物资源持续利用国家重点实验室,昆明650201;2、中国科学院大学,北京100049;3、云南大学,化学科学与工程学院,昆明650091)
结合核磁共振技术与液质联用技术的代谢组学数据采集、处理和分析
李兴1,2,余玲玲1,3,胡凯锋1*
(1、中国科学院昆明植物研究所植物化学与西部植物资源持续利用国家重点实验室,昆明650201;2、中国科学院大学,北京100049;3、云南大学,化学科学与工程学院,昆明650091)

胡凯锋博士,1995年毕业于北京医科大学药学院,1998年获中国科学院上海药物所硕士,此后分别在美国马里兰大学和瑞士联邦苏黎世高等理工学院学习,2004年获得博士学位。2005年起在美国国立卫生研究院(NIH)任研究助理,2009年至2012年任职于美国威斯康星大学,期间主要负责建立了基于结合液质联用和核磁共振技术的代谢组学研究平台,2012年入选国家“青年千人计划”。同年加盟中国科学院昆明植物研究所,任研究员、博士生导师,组建“天然药物药效物质基础与作用机制”研究组,并负责研究所液态核磁共振中心的建设。开发了一些列的核磁共振技术定性、定量方法,其中对复杂混合物体系中单体进行绝对定量的二维核磁共振方法,HSQC0被广泛用于天然药物化学、代谢组学研究中。
摘要:代谢组学是近年来新兴起来的一门“组学”学科,主要研究不同状态下,如正常与应激、健康与疾病、野生型与基因突变型,生物系统内所有小分子代谢物(M.W.< 1500 Da)的变化,对其进行定性鉴定和定量分析,目的是发现和鉴别差异代谢物,揭示差异代谢物在不同状态下生物系统中的产生途径及其作用。代谢组学研究通常包括样品制备、数据采集、数据(预)处理和多变量分析等步骤,正确的数据采集、处理和分析是获得具有统计意义结果的前提。核磁共振技术(nuclear magnetic resonance, NMR)和液质联用技术(liquid chromatography-mass spectroscopy,LC-MS)是代谢组学研究的两种最主要分析手段。本文将主要针对结合NMR和LC-MS两种技术进行代谢组学研究的数据采集、处理、分析进行综述,并展望NMR和LC-MS两种技术在代谢组学领域的发展前景。
关键词:代谢组学;核磁共振技术;液质联用技术;数据采集;数据处理;数据分析
1 前言
代谢组学是系统生物学的一个分支,常作为发现代谢标志物的一种工具。近年来随着生物信息学和分析技术的发展及多种正交生物学方法的整合,代谢组学分析已从发现生物标志物扩展到理解其在生物系统中的作用。代谢物作为底物参与驱动细胞本质生命活动,如能量产生和储存、信号转导和细胞凋亡等过程,因而代谢组学所检测到的微小变化可为理解不同生理或病理机制提供另一种新的视角[1]。代谢组学已广泛应用于医学、药学、生物学、植物学、食品分析及环境分析等各个领域。核磁共振技术(NMR)和液质联用技术(LC-MS)是代谢组学研究的两种主要分析工具,它们相互独立并具有一定技术互补性。NMR可提供丰富的结构信息(如化学位移、耦合常数、空间相互作用)能够用于代谢物的定性鉴别,此外,NMR图谱信号强度直接正比于磁性原子核的数目,使得它可以作为一种普适性的定量分析工具[2]。利用NMR技术进行代谢组学研究还具有样品制备简单、数据采集快速(1D NMR谱通常只要6-10 min)、无损分析等特点。NMR技术的主要弱点在于因NMR能级裂分较小而表现出较低的灵敏度,一般比LC-MS低3个数量级;复杂混合物体系(代谢组) 1D NMR谱的分辨率有限,常出现信号峰重叠严重;2D或nD NMR实验时间通常较长。LC-MS 结合色谱高效分离与质谱高检测灵敏度的特性,使其成为代谢组学研究的另一种主要分析工具[3]。除了色谱维度的保留时间能够一定程度上表征化合物的化学性质,高分辨质谱还可提供代谢物的精确分子量及同位素峰型等信息,而且二级或多级质谱的裂解片段信息均可帮助推断代谢物的结构[4]。利用同位素内标或外标浓度工作曲线为参照,LC-MS可以进行定量、半定量分析以及代谢指纹图谱分析。虽然LC-MS具有高效分离与高灵敏度检测的优点,但一些生物活性物质或生物合成中间体具有化学不稳定性,容易在色谱分离过程中降解,而且质谱的离子化程度容易受化合物本身的性质以及基质效应等多种因素影响。因此,利用NMR技术(包含丰富的结构信息以及定量无偏性的特点)和LC-MS技术(具有高效分离与高检测灵敏度的特性)良好的互补性,近年来,结合这两种重要分析技术在代谢组学研究中的应用变得越来越广泛[5,6]。本文主要探讨NMR和LC-MS技术正确的数据采集、处理和分析方法对于获得可靠的、具有统计学和生物学意义的差异代谢物的重要性,并对这两种技术在代谢组学研究中的发展趋势进行总结和展望。
2 数据采集
2.1NMR数据采集
由于氢元素普遍存在于绝大多数的代谢物中且氢核磁共振(1H NMR)具有较好的灵敏度,本文将主要以1D1H NMR的数据采集为例,介绍采样参数的设置和脉冲序列的选择。但下面介绍的原理对其它杂核的1D和2D NMR数据采集同样具有参考价值。
核磁数据点的采集频率1/Δt (Δt为dwell time)必须满足大于或等于感兴趣的代谢物化学位移谱宽区域。对于复杂混合物体系,如代谢组样品,为了提高NMR信号在频率域的分辨率、减少谱峰重叠,一般尽可能延长时间域数据采集时间(tmax)。然而当采样时间过长时,信号由于横向弛豫衰减可能低于噪音水平,所获得的数据将包含相对更多的噪音,从而导致信号噪音比(signal-noise ratio, SNR)降低。一般采样时间设在0.75-1.26 T2范围内,在获得较高分辨率的同时不牺牲灵敏度[7]。在nD NMR实验中[7,8],采用非均匀采样(non-uniform sampling, NUS)策略可以在单位采样时间内获得较高的图谱分辨率。
用于代谢组学研究的核磁共振谱图具有一定程度的相对定量特性,对于复杂代谢物混合物,其化学位移涵盖的谱宽区域相对较宽,一般采样时尽可能将频率偏置中心(offset, O1值)设置在代谢物混合物化学位移的中心位置以减少由于偏置效应对核磁信号强度不同程度的影响。用于定量代谢组学研究的1D1H qNMR (quantitative NMR),多次扫描实验的扫描时间间隔 (interscan delay),d1要求 ≥ 5T1max(T1max为代谢物混合物的所有核磁信号最长的纵向弛豫时间)从而使得代谢物混合物的所有核磁信号能够最大程度(>99%)恢复到起始的平衡态,平衡态下代谢物核磁信号的起始磁化矢量与该代谢物的浓度直接成正比。高通量核磁数据采集过程中常采用较短的扫描时间间隔(<< 5T1max),但只要样品间采用相同的扫描时间间隔,即可认为磁化矢量获得近似相同程度的平衡态恢复,从而不影响样品间特定代谢物相对量的比较。
对于复杂代谢物混合物,比如含有蛋白质的代谢组样品,可以应用图谱编辑(spectral editing) 技术简化1D1H NMR图谱,以减少信号重叠和背景信号对基线以及信号峰积分的影响,从而提高1D1H NMR图谱定性、定量的可靠性。由于生物大分子(蛋白质分子)的横向弛豫(T2relaxation, T2r)时间与代谢物小分子相比通常较短,一种较为常见的1D1H NMR图谱编辑方法是通过在激发脉冲后插入一段CPMG (Carr-Purcell-Meiboom-Gill)脉冲序列,利用生物大分子较快的横向弛豫实现对其核磁共振信号的压制过滤。如果混合物中代谢物分子是自由的,即假设认为代谢组样品内不存在影响代谢物横向弛豫T2r的分子间相互作用(如代谢物与蛋白质分子间的结合),应用CPMG脉冲序列压制生物大分子的核磁共振信号,通常对代谢物本身的核磁信号强度的影响较小。但如果代谢物分子与样品内蛋白分子间存在相互作用,处于结合态的代谢物分子的核磁信号在CPMG脉冲过滤中会较大程度的衰减,且某种代谢物核磁信号损失的比例会受其所处的代谢组样品条件的影响(称为基质效应1,如样品中包含不同量残留的蛋白质、残留蛋白的分子量以及蛋白质分子与该代谢物分子间相互作用的强弱等)。因此代谢物与样品中背景物质的相互作用(基质效应1)可能会影响样品间该代谢物的相对定量。一般在样品处理阶段可以利用甲醇或其他溶剂沉淀除掉蛋白质及其他生物大分子。
此外,代谢组样品间盐浓度(基质效应2)的不同会从两个方面影响样品间代谢物的相对定量。其一,样品盐浓度的不同通常会影响90o脉冲宽度,具有不同盐浓度的代谢组样品(比如尿液代谢组样品),如果平行高通量数据采集过程中使用相同的激发脉冲长度,由于盐浓度对每个实际样品90o脉冲宽度的影响(基质效应2)从而导致实际磁化矢量受激发程度的不同而影响样品间代谢物的相对定量。此外,90o脉冲宽度的不同也会对偏置效应产生一定的影响从而影响定量。其二,根据相互原则(principle of reciprocity)[9],不同盐浓度(基质效应2)会影响样品溶液的介电常数,从而影响待检测的磁化矢量与探头线圈之间的耦合,即影响探头线圈的信号接受效率,导致线圈中诱导产生的电流(free induction decay, FID)不同,获得信号强度需要根据PULCON (pulse length based concentration determination)原理进行校正。因此,对于基质差异(基质效应1和2,即样品基质中的蛋白质和盐浓度差异)较大的代谢组样品,不应过分依赖自动化的高通量核磁数据采集方法(比如设置相同的90o脉冲宽度),谨慎地应用各种图谱编辑技术,以获得更可靠的,能够更真实反映代谢组样品组成的核磁数据。当然,可以通过样品前处理步骤尽可能去除基质的差异。
此外,代谢组学经常应用1D1H NOESY实验采集NMR数据。通常在1D1H NOESY(nuclear Overhauser effect spectroscopy)脉冲序列的开始采用一段长时、低功率的连续波照射对溶剂信号进行预饱和,从而能选择性抑制溶剂峰信号[10],因此该方法适用于溶剂质子化浓度较高的代谢组学样品,比如水相代谢组样品(如血浆、血清、尿液)。为克服1D1H NMR谱的信号峰重叠问题,近年来2D NMR脉冲序列如HSQC(heteronuclear single-quantum coherence)、COSY(correlation spectroscopy)、TOCSY(total correlation spectroscopy)等也被广泛用于代谢组学分析[2]。
2.2LC-MS数据采集
基于LC-MS的代谢组学数据采集涉及色谱分离和质谱检测两方面因素。在代谢组学研究中通常采用超高效液相色谱(ultra performance liquid chromatography, UPLC)系统结合不同性质的色谱柱的策略,以提高色谱分离效率、扩大对代谢物的覆盖范围。与传统HPLC(high performance liquid chromatography)相比,UPLC色谱柱填料粒径小(< 2 μm),柱效高,分析时间短,可提高分离度,增加分辨率和峰容量,通过高效分离减轻基质效应对质谱离子化的影响,提高样品分析通量。为获得全面的代谢物谱,在LC-MS代谢组实验中常结合使用反相色谱柱(如C18柱)和亲水作用色谱(hydrophilic interaction liquid chromatography, HILIC)柱对复杂生物样品进行分离[11,12]。两者在选择性方面具有互补性,前者可分离极性较小的物质,后者主要分离极性物质(主要为初级代谢物)。
质谱检测方面主要考虑三个关键因素:离子源、质量分析器和二级质谱(MS/MS)采集方法。为增加代谢物覆盖范围,一般常采用大气压离子化技术,如电喷雾离子化(electrospray ionization, ESI)、大气压化学电离(atmospheric pressure chemical ionization, APCI)、大气压光致电离(atmospheric pressure photoionization, APPI)分别在正、负离子模式下采集数据。ESI是一种软电离方式,可获得完整的分子离子,帮助代谢物的初步鉴别。与ESI类似,APPI和APCI也很少或几乎不源内裂解形成离子片段, APPI和APCI离子源可耐受相对高浓度的缓冲液,常作为ESI的替代/补充方式用于检测低极性、热稳定性化合物(如脂质)[12]。一种新的趋势是采用ESI/ APCI或ESI/APPI组合成的单一离子源配置。在优化离子源的同时,不同功能的质量分析器常通过串联或杂合配置用于采集高分辨率和高准确度的MS/MS质谱,进一步对代谢物进行识别和结构确证。常见的质量分析器组合方式包括:基于四极杆的串联质谱,如QqQ (triple quadrupole)或QTOF(quadrupole time-of-fl ight);基于离子肼的串联质谱,如QIT(quadrupole ion trap)或LIT(linear ion trap)-Orbitrap[12]。由于QqQ在多反应监测模式(multiple reaction monitoring, MRM)下的高选择性和高专属性,它常作为小分子化合物绝对定量的分析模式。MS/MS数据采集主要分为两类[13]:数据依赖性采集(data dependent acquisition, DDA)和数据非依赖性采集(data independent acquisition, DIA)。DDA也称信息依赖采集,指根据预定义的前体离子选择标准,先完成一轮MS全扫描并自动选择前体离子,随后对前体离子依次进行MS/MS扫描。前体离子选择标准主要包括离子强度、准确质量包含列表等。DIA是一种基于非选择性碰撞诱导解离(collision-induced dissociation, CID)的方法,它是对一个m/z窗口范围内的所有离子采集MS/MS而非选择单个前体离子。这种数据采集方式首先在QTOF仪器(梯度碰撞能量采集方法MSE)上实现并应用于代谢物鉴别,随后Orbitrap(“所有离子片段”方法)和TripleTOF® (MS/MSALLwith SWATHTMacquisition)系统也采用这种策略。目前这种数据采集策略可与HPLC结合使用,受扫描速度制约还不能与UPLC完全兼容。
3 数据处理
3.1NMR代谢组学数据处理
从原始FID数据开始,常规的NMR数据处理包含以下步骤:加窗函数、填零、傅里叶变换、相位校正、定标、基线校正和背景扣除(见图1(A)),用于代谢组学研究的NMR数据处理步骤还包括:信噪比计算、峰识别(peak dection)、拟合/去卷积(fitting/deconvolution)、分段(binning/bucketing)积分或峰积分(integration)、匹配(matching)与对齐(alignment)、等步骤(见图1(B))。本文将主要介绍其中与代谢组学研究密切相关又至关重要的数据处理步骤:峰识别、峰积分与分段积分、匹配与对齐。
3.1.1峰识别
谱峰的识别一般可以基于峰型识别或峰强度识别或者采取两者相结合的峰识别方式。峰型识别可以通过计算局部范围内信号强度的变化率(一阶导数),设定一定的阈值用于识别信号峰及判断谱峰的坐标位置(如二阶导数为0)。峰强度识别通常基于信号噪音比大于设定的阈值,噪音水平可以选择一段没有任何信号的区域计算获得。峰的坐标位置可以通过局部峰型拟合、外推法或权重平均计算获得。
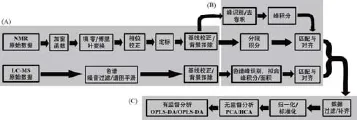
图1. NMR-和LC-MS-代谢组学数据处理与数据分析流程示意图.
3.1.2峰积分与分段积分
由于高分辨1D或 2D NMR图谱通常包含较多的数据点(独立变量),在代谢组学多变量分析前需要对原始NMR图谱数据进行降维处理,通常可对图谱进行谱峰识别、峰积分或采用分段积分处理。谱峰的积分范围(信号峰的起止点)可以根据半峰宽进行确定。
分段积分是将谱图划分为等间隔或不同间隔的区间,计算每段的积分值用以表示图谱的变量值,这种处理方式可规避某些小(间隔范围内)的谱峰漂移误差。分段积分实质是对一定间隔范围内信号进行加和从而减少图谱数据点(独立变量)的数目实现降维,因而同时谱图的分辨率降低。谱图分段积分算法有多种,较为常见的,比如AMIX(analysis of mixtures, Bruker)软件采用等间隔(0.04 ppm)分段积分算法。这种分段积分算法将代谢组学每个样品的谱图划分为完全相同的等间距的积分区间[14],一个明显缺点在于这种分段法无从区分感兴趣的信号峰区域和没有任何信号的噪音区域,缺乏分段边界定义的灵活性,因而可能造成对大量毫无意义的噪音数据进行积分或信号峰被划分在两段积分区间内。为防止信号峰被等间隔分段的边界拆分,一些非等间距分段积分的算法被提出,如自适应分段积分(adaptive binning)[15]、高斯分段积分(Gaussian binning)[16]、自适应智能分段积分(adaptive intelligent binning, AI-binning)[17]、动态自适应分段积分(dynamic adaptive binning)[18],这些算法的优点在于:考虑信号峰的位置,通过智能化灵活定义分段边界避免信号峰被拆分。
3.1.3峰匹配与对齐
由于仪器、pH值、温度、盐浓度、样品稀释度等因素均可造成NMR谱峰的化学位移漂移现象,为获得对代谢物的正确鉴别和准确定量,样品间谱峰的匹配与对齐是进行代谢组学多变量分析前非常重要的数据处理步骤。对于化学位移的系统性误差, 一般可采用内标(如TMS(tetramethylsilane),TSP(sodium 2,2-tetradeutero-3-trimethyl-silylpropionate)等)对谱图进行化学位移校正(也称定标)。但对复杂的代谢组混合物样品,由于样品间的物质组成、pH值、盐浓度、样品稀释度等不完全一致以及采样温度的波动等因素,特定代谢物受样品中复杂背景物质的影响(基质效应)其化学位移在代谢组样品间的漂移往往是非全局性的[19],因此在内标校正的基础上,还需要对样品间的谱峰进行匹配和化学位移漂移的局部对齐。样品间谱峰的正确匹配能够帮助代谢物的鉴别,谱峰匹配的逻辑算法主要基于代谢物信号峰的绝对与相对位置(化学位移与信号峰模式)、不同信号峰强度的相对比例以及峰型。常用的NMR谱峰对齐算法有icoshift(interval correlation optimised Shifting)[20]和COW(correlation optimized warping)[21-22],它们都是基于区间局部对齐的算法。Icoshift算法将谱图划分为不同长度的区间,根据特定区间局部的相关性采用循环移位(circular shift)的方式与参照谱图的相应区间进行对齐。COW算法是将谱图划分为等长度区间,通过延伸或压缩区间的方式与参照谱图的相应区间规整对齐。COW算法源自色谱谱峰的对齐,目前已成功用于NMR谱峰的对齐[23]。
3.2LC-MS代谢组学数据处理
LC-MS数据是由保留时间与质荷比构成的二维变量空间的信号强度数据集[24]。由于同位素峰、不同加合离子状态及中性丢失的存在,为了能有效挖掘反映生物表型差异的生物标志物,需对数据进行一系列前处理操作,包括:质谱维同位素峰去除及不同加合离子和碎片离子集成,色谱维噪音过滤/谱图平滑、基线校正和背景扣除、信噪比计算,峰识别、拟合/去卷积(重叠峰解析)获得峰积分或峰面积、峰匹配与对齐等(见图1)。目前研究者已开发出许多相关的数据处理软件,主要可概括为3类[25,26]:商业软件,如MarkerView (AB Sciex), MarkerLyn(Waters), MassProfi ler Professional (Agilent),SIEVE (Thermo),ProfileAnalysis (Bruker)和MassHunter (Agilent)等;免费软件如MZmine[27]、XCMS[28]and MetAlign[29]等;和一些用Matlab、R语言编写的脚本程序。已有不少关于LC-MS数据处理的相关综述报道[12,24],本文将主要关注LC-MS数据处理两个重要关键步骤:谱峰识别、峰匹配与保留时间对齐。
3.2.1色谱峰识别
峰识别有多种逻辑算法,如连续小波变换处理、基于模型(如高斯函数)的峰拟合或匹配过滤等[30]。峰识别通常主要基于以下标准,如信号噪音比(SNR),强度阈值(detection/ intensity threshold)、峰斜率(slopes of peaks, 一阶导数)、局部极大值(local maximum)、峰形比值(shape ratio)、脊线(ridge lines, 小波变换处理)、拟合峰宽(peak width)。例如,Du等人[31]提出基于连续小波变换处理,将小波空间的脊线定义为峰。基于模型的峰拟合或匹配过滤算法可以确定峰的起止范围、峰积分/面积和峰位置;积分范围(信号峰的起止点)可以基于半峰宽或者匹配峰型函数(如高斯函数)的拐点确定。
3.2.2峰匹配与保留时间对齐
保留时间RT(retention time)或m/z系统性误差(整体漂移)可用单调函数进行拟合校正,而样品组分特异性的RT或m/z的局部漂移则需要通过局部峰匹配与非线性校正对齐。用于处理LC-MS保留时间漂移问题的峰对齐算法有很多种[32],较常见的,比如采用warping函数规整校正样品组分特异性的RT非线性漂移。聚类warping方法对于有信号缺失的样品分析具有稳健性,因为它会对全部代谢组样品的所有特征进行组合和聚类,而不依赖于原始单个样品的数据。动态时间规整算法(dynamic time warping,DTW)将一定质量范围内(0.25 m/z)的多个离子色谱峰用较宽 (standard deviation, SD较大) 的高斯函数进行平滑处理,使保留时间粗略(较大的保留时间区间内)相似的色谱峰匹配组合成峰簇,平滑处理变换成虚拟的基元峰(meta-peak, 用其代表一组保留时间相近的色谱峰)。先将样品间基元峰进行匹配对齐(相当于整体轮廓性匹配),并利用其中最强的组分峰进行保留时间局部非线性校正对齐。逐步缩小用于平滑处理的高斯函数峰宽进行更精确的匹配组合,通过多次的匹配、对齐迭代实现对保留时间越来越精确的非线性校正对齐。
COW[21]时间规整算法类似于DTW,不同之处在于COW是基于色谱峰的相关性进行保留时间规整对齐,而DTW是基于色谱峰的分布进行保留时间规整对齐,两者可以互为补充。除warping方法外,还可以基于峰特征(如精确分子量、保留时间、同位素峰型、二级质谱特征)的相似性进行直接峰匹配再对保留时间进行非线性校正对齐,如RTAlign[33]、Peakmatch[34]等。虽然报道了许多用于LC-MS数据对齐的算法,但针对一些问题仍有待进一步优化完善[32],如:拟合色谱峰模型能否反映真实的RT漂移;算法复杂性及用户定义优化参数导致数据处理计算的时间成本增加;大多数对齐算法之间缺少对比评估。
4 数据分析
无论是NMR还是LC-MS数据,经过前处理匹配与对齐的数据表格,在进行多变量分析之前通常还需要经过数据过滤或补齐(data filter/refill)、归一化(normalization)、均值中心化和标准化(scaling)等预处理步骤(见图1(C)),有时根据数据的分布,还需要对原始数据进行对数转换和指数转换处理。代谢组学数据多变量分析主要分为:无监督模式多变量分析(如主成分分析(principal component analysis, PCA)、层级聚类分析(hierarchical cluster analysis, HCA)等)和有监督模式多变量分析(如偏最小方差判别分析(partial least square discriminant analysis, PLS-DA)、正交偏最小方差分析(orthogonal partial least square discriminant analysis, OPLS-DA)等)。PCA和PLS是代谢组学数据分析最常用的两种多变量分析方法,PCA分析是一种无监督的降维分析方法,用于寻找原始变量空间样品间变化差异较大的主要变量(即主要成分),而PLS-DA分析属于有监督分析,其实质是一种降维判别分析方法,通过投影(降维)处理,在原始变量空间寻找能够最好进行样品组间区分的空间变量(即原始代谢组数据在该变量方向上的得分值与组间区分具有最好的相关性),再通过负载分析获得与组间区分(判别)相关的原始变量。目前已有多种数据分析软件如MVAPACK、SIMCA-P、Unscrambler等可实现多变量分析,其中SIMCA-P使用最广泛[35]。此外,不同来源的代谢组数据既可分别进行多变量分析也可经过数据融合后再进行多变量分析,数据融合分析有利于增强模型的分类及预测能力。对多变量分析结果的解释应考虑数据的预处理过程,有时需要进行回溯转换以便更好地解释原始数据的生物学意义。此外,代谢组学数据分析可以采用多变量分析与单变量分析(如T-检验、U-检验、方差分析等)相结合的策略获得更可靠的结果。
5 结束语
NMR和LC-MS作为代谢组学研究最常用的两种分析技术,具有一定的技术和信息互补性,同时它们作为两种相互独立、正交的技术手段,其统计分析结果可以相互佐证加强。除了NMR和LC-MS两种来源的数据可以进行融合分析外,还可以结合其他来源的数据(如GC-MS、MIR、NIR数据等)或其他组学 (基因组、转录组、蛋白组)数据进行综合分析,这是未来生命科学研究中寻求合理解释许多生命现象的一种发展趋势。正确的数据采集方法、恰当的数据处理方法和合理的数据分析方法对于获得可靠的、具有统计学意义和生物学意义的结果,深入理解不同状态(如健康与疾病、正常与应激、野生型和突变型)生物系统的潜在机制至关重要。目前基于NMR和LC-MS代谢组学数据的采集、处理和分析仍存在许多亟待进一步优化和完善的方面:(1)多维NMR实验时间较长,制约其在高通量分析大规模样本中的应用;(2)如何进一步提高数据采集、处理、分析的自动化程度;(3)用于代谢组学研究的各种数据处理、分析算法和软件所获得的结果的真实性和合理性评估,各种数据处理、分析算法和软件之间的对比评估以及各自的适用范围以及优缺点等。
参考文献
[1] Johnson C H, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms[J]. Nat Rev Mol Cell Biol, 2016, doi:10.1038/nrm.2016.25.
[2] Bingol K, Bruschweiler R. Multidimensional approaches to NMR-based metabolomics[J]. Anal Chem, 2014, 86(1): 47-57.
[3] Gika H G, Wilson I D, Theodoridis G A. LC-MS-based holistic metabolic profiling. Problems, limitations, advantages,and future perspectives[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2014, 966: 1-6.
[4] 孔宏伟, 戴伟东, 许国旺. 基于液相色谱-质谱联用的代谢组学研究中代谢物的结构鉴定进展[J]. 色谱, 2014, 32(10): 1052-1057.
[5] Johanningsmeier S D, Harris G K, Klevorn C M. Metabolomic technologies for improving the quality of food: practice and promise[J]. Annu Rev Food Sci Technol, 2016, 7: 413-438.
[6] Spiteri M, Dubin E, Cotton J, et al. Data fusion between high resolution 1H-NMR and mass spectrometry: a synergetic approach to honey botanical origin characterization[J]. Anal Bioanal Chem, 2016, 408(16): 4389-4401.
[7] Rovnyak D, Sarcone M, Jiang Z. Sensitivity enhancement for maximally resolved two-dimensional NMR by nonuniform sampling[J]. Magn Reson Chem, 2011, 49(8): 483-491.
[8] Mobli M, Hoch J C. Nonuniform sampling and non-Fourier signal processing methods in multidimensional NMR[J]. Prog Nucl Magn Reson Spectrosc, 2014, 83: 21-41.
[9] Wider G, Dreier L. Measuring protein concentrations by NMR spectroscopy[J]. J Am Chem Soc, 2006, 128(8): 2571-2576.
[10] McKay R T. How the 1D-NOESY suppresses solvent signal in metabonomics NMR spectroscopy: an examination of the pulse sequence components and evolution[J]. Concepts Magn Reson Part A, 2011, 38A(5): 197-220.
[11] Theodoridis G A, Gika H G, Want E J, et al. Liquid chromatography-mass spectrometry based global metabolite profi ling: A review[J]. Anal Chim Acta, 2012, 711: 7-16.
[12] Xiao J F, Zhou B, Ressom H W. Metabolite identifi cation and quantitation in LC-MS/MS-based metabolomics[J]. Trends Analyt Chem, 2012, 32: 1-14.
[13] Ma S G, Chowdhury S K. Data acquisition and data mining techniques for metabolite identifi cation using LC coupled to high-resolution MS[J]. Bioanalysis, 2013, 5(10): 1285-1297.
[14] Izquierdo G J L, Villa P, Kyriazis A, et al. Descriptive review of current NMR-based metabolomic data analysis packages[J]. Prog Nucl Magn Reson Spectrosc, 2011, 59(3): 263-270.
[15] Davis R A, Charlton A J, Godward J, et al. Adaptive binning: an improved binning method for metabolomics data using the undecimated wavelet transform[J]. Chemometr IntellLab Syst, 2007, 85(1): 144-154.
[16] Anderson P E, Reo N V, DelRaso N J, et al. Gaussian binning: a new kernel-based method for processing NMRspectroscopic data for metabolomics[J]. Metabolomics,2008, 4(3): 261-272.
[17] De M T, Sinnaeve D, Van Gasse B, et al. NMR-based characterization of metabolic alterations in hypertension using an adaptive, intelligent binning algorithm[J]. Anal Chem, 2008,80(10): 3783-3790.
[18] Anderson P E, Mahle D A, Doom T E, et al. Dynamic adaptive binning: an improved quantifi cation technique for NMR spectroscopic data[J]. Metabolomics, 2011, 7(2): 179-190.
[19] Giskeodegard G F, Bloemberg T G, Postma G, et al. Alignment of high resolution magic angle spinning magnetic resonance spectra using warping methods[J]. Anal Chim Acta,2010, 683(1): 1-11.
[20] Savorani F, Tomasi G, Engelsen S B. Icoshift: a versatile tool for the rapid alignment of 1D NMR spectra[J]. J Magn Reson,2010, 202(2): 190-202.
[21] Nielsen N P V, Carstensen J M, Smedsgaard J. Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimised warping[J]. J Chromatogr A, 1998, 805(1-2): 17-35.
[22] Tomasi G, Van D B F, Andersson C. Correlation optimized warping and dynamic time warping as preprocessing methods for chromatographic data[J]. J Chemom, 2004, 18(5): 231-241.
[23] Rosenling T, Stoop M P, Smolinska A, et al. The impact of delayed storage on the measured proteome and metabolome of human cerebrospinal fl uid[J]. Clin Chem, 2011, 57(12): 1703-1711.
[24] Gika H G, Theodoridis G A, Plumb R S, et al. Current practice of liquid chromatography-mass spectrometry in metabolomics and metabonomics[J]. J Pharm Biomed Anal, 2014,87: 12-25.
[25] 汪明明, 程海婷, 薛明. 基于 LC-MS 的代谢组学分析流程与技术方法[J]. 国际药学研究杂志, 2011, 38(2): 130-136.
[26] Rafiei A, Sleno L. Comparison of peak-picking workfl ows for untargeted liquid chromatography/ high-resolution mass spectrometry metabolomics data analysis[J]. Rapid Commun Mass Spectrom, 2015, 29(1): 119-127.
[27] Katajamaa M, Miettinen J, Oresic M. MZmine: toolbofor processing and visualization of mass spectrometry based molecular profi le data[J]. Bioinformatics, 2006, 22(5): 634-636.
[28] Mahieu N G, Genenbacher J L, Patti G J. A roadmap for the XCMS family of software solutions in metabolomics[J]. Curr Opin Chem Biol, 2016, 30: 87-93.
[29] Lommen A. MetAlign: interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing[J]. Anal Chem, 2009, 81(8): 3079-3086.
[30] Yang C, He Z Y, Yu W C. Comparison of public peak detection algorithms for MALDI mass spectrometry data analysis[J]. BMC Bioinformatics, 2009, 10: 4.
[31] Du P, Kibbe W A, Lin S M. Improved peak detection in mass spectrum by incorporating continuous wavelet transformbased pattern matching[J]. Bioinformatics, 2006, 22(17): 2059-2065.
[32] Smith R, Ventura D, Prince J T. LC-MS alignment in theory and practice: a comprehensive algorithmic review[J]. Brief Bioinform, 2013, 16(1): 104-117.
[33] Duran A L, Yang J, Wang L J, et al. Metabolomics spectral formatting, alignment and conversion tools (MSFACTs)[J]. Bioinformatics, 2003, 19(17): 2283-2293.
[34] Johnson K J, Wright B W, Jarman K H, et al. Highspeed peak matching algorithm for retention time alignment of gas chromatographic data for chemometric analysis[J]. J Chromatogr A, 2003, 996(1-2): 141-155.
[35] Triba M N, Le Moyec L, Amathieu R, et al. PLS/OPLS models in metabolomics: the impact of permutation of dataset rows on the K-fold cross-validation quality parameters[J]. Mol Biosyst Biosys, 2015, 11(1): 13-19.
Email: kaifenghu@mail.kib.ac.cn
中图分类号:Q591
文献标识码:A
DOI:[CLC Number] Q591[DocumentCode] A10. 11967/ 2016140301 10.11967/2016140301
基金项目:⋆国家自然科学基金项目(21505142),云南省高端科技人才引进计划(2012HA015),青年千人计划
作者简介:李兴,男,博士研究生,研究方向:细胞代谢组学
通讯作者:胡凯锋,男,研究员
Data Acquisition, Processing and Analysis for NMR- and LC-MS-based Metabolomics
Xing Li1,2, Lingling Yu1,3, Kaifeng Hu1*
( 1、State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, P. R. China;2、University of Chinese Academy of Sciences,Beijing100049, P.R. China; 3、School of Chemical Science and Technology, Yunnan University, Kunming 650091, P. R. China )
Abstract:Metabolomics has been becoming a booming and widespread '-omics' technique over the past decades, which qualitatively and quantitatively analyzes the changes of all small molecule metabolites(M. W. < 1500 Da)in a biological system ?under different states, such as normal and stress, health and disease, of wild and mutant type, aiming to discover and identify differential metabolites, as well as to reveal their synthetic pathways and biological roles. A workfl ow of metabolomics studies generally comprises sample preparation, data acquisition, data(pre)processing and data analysis. Proper data acquisition,processing and analysis are the prerequisites for obtaining statistically significant and meaningful results. Nuclear magnetic resonance(NMR)and liquid chromatography-mass spectroscopy(LC-MS)are two predominant analytical platforms for metabolomics study. Here, we review the data acquisition, processing and analysis for metabolomics study using NMR and LCMS technologies, as well as their prospects.
Key Words:Metabolomics; NMR; LC-MS; Data acquisition; Data processing; Data analysis
