玉米抗禾谷镰刀菌的转录组分析
2016-08-27刘永杰马传禹马雪娜徐明良
刘永杰 马传禹 马雪娜 徐明良
中国农业大学国家玉米改良中心, 北京100193
玉米抗禾谷镰刀菌的转录组分析
刘永杰 马传禹 马雪娜 徐明良*
中国农业大学国家玉米改良中心, 北京100193
赤霉菌茎腐病是由禾谷镰刀菌(Fusarium graminearum, 有性态, Gibberella zeae)引起的一类土传性病害, 严重危害玉米的产量和品质。本研究依据玉米第10和第1染色体上的2个抗茎腐病QTL, qRfg1和qRfg2、培育近等基因系NIL-R (2个QTL位点均为抗病等位基因)和NIL-S (2个QTL位点均为感病等位基因)。在成株期和幼苗期接种禾谷镰刀菌, 两近等基因系的抗性差异均显著。用2个近等基因系的幼根接种禾谷镰刀菌, 进行转录组分析研究。结果表明, 与NIL-S相比, NIL-R在接种禾谷镰刀菌后, 乙烯(ethylene, ET)合成、信号途径基因, 病程相关蛋白、脱氧雪腐镰刀菌烯醇毒素(deoxynivalenol, DON)解毒基因等呈现特异上调表达。与NIL-S相比, 有1170个基因在NIL-R对照组中表达量较高, 其中水杨酸(salicylic acid, SA)、茉莉酸(jasmonic acid, JA)和乙烯合成和信号介导途径以及苯丙烷合成途径中的基因显著富集; 接种禾谷镰刀菌6 h或18 h后, 病程相关蛋白、激素JA和ET合成基因、DON解毒基因在NIL-R中表达量较高。
玉米; 茎腐病; 转录组; 抗性; JA/ET; 苯丙烷
玉米茎腐病, 又称茎基腐病, 是一类危害严重的土传性病害, 降低玉米的产量和品质。茎腐病由多种病原菌通过根系或茎基部伤口单独或复合侵染玉米所致, 其中包括腐霉菌、炭疽菌和镰刀菌等。赤霉菌茎腐病是由禾谷镰刀菌(Fusarium graminearum, 有性态Gibberella zeae)引起[1]。该病原菌还可引起玉米穗腐病[2], 小麦[3]和大麦[4]赤霉病。此外,禾谷镰刀菌还可以侵染水稻[5]和拟南芥[6]。
禾谷镰刀菌是镰刀属致病菌的一种, 该类病菌分泌的脱氧雪腐镰刀菌烯醇毒素(deoxynivalenol,DON), 严重危害人畜的健康; 美国食品药品委员会规定实用面粉中该毒素的含量不能超过1 µg g-1[7]。DON对细胞功能的影响在动物中研究较为深入, 有报道称DON可与60S核糖体亚基结合来抑制蛋白质的合成, 进而激活细胞衰亡程序导致细胞死亡[8-10]。植物中, DON可体外抑制小麦核糖体中蛋白的合成[11]。DON并非病原菌生长必需, 但有报道称该毒素影响病原菌致病性, DON可作为一种有毒因子参与到病原菌侵染寄主的过程中[10]。与野生型相比, 毒性合成基因tri-5突变株在侵染小麦和玉米时致病力减弱[12-14];DON不仅可引起细胞内过氧化氢的积累以至于引起细胞程序化死亡, 还可以激活抗性响应基因病程相关蛋白(PR)的表达[15]。寄主细胞可通过诱导毒素降解基因或毒素转运基因的表达来降解毒素或将毒素转运出胞质[16]。小麦抗赤霉病 QTL-Fhb1可能编码一种DON-葡糖基转移酶(DON-glucosyltransferase,DOGT)或者能够调节该转移酶的蛋白[17]。有报道指出UDP-葡糖基转移酶(UDP-glucosyltransferases, UGTs)和ABC转运蛋白(ABC transporters)能够降低DON的毒性[17-18]。DON或镰刀菌接种的寄主中都会诱导产生解毒蛋白如谷胱甘肽 S-转移酶(glutathione S-transferases, GSTs)、多药抗性蛋白 multidrug resistance protein (MRP)、MATE efflux family proteins、重金属转运蛋白(heavy metal transport/detoxification proteins)、多向耐药性蛋白(pleiotropic drug resistance,PDR)、PDR类 ABC转运蛋白(PDR type ABC transporters)等[19-21]。
Kruger等[22]将禾谷镰刀菌接种到小麦颖壳后发现 312个基因差异表达, 其中包括编码苯丙烷代谢途径的酶类、氧化还原过程酶类、GSTs、PR蛋白和其他抗性或逆境相关的蛋白的基因; Boddu等[20]利用DON接种大麦后发现被诱导的主要为PR蛋白基因、氧化胁迫相关基因、苯丙烷类合成酶。Zhu等[23]以 F. oxysporum接种拟南芥后发现多种植物激素,茉莉酸(jasmonic acid, JA)、乙烯(ethylene, ET)、水杨酸(salicylic acid, SA)、生长素(auxin)及信号传导途径基因都被诱导。Lanubile等[24]利用 Fusarium verticillioides接种穗腐病抗性不同的玉米后发现, 编码PR蛋白、苯丙烷代谢途径酶类的基因、氧化胁迫相关基因、ET信号途径的基因在抗病和感病材料中都被诱导, JA信号途径在抗病材料CO441中被诱导。
尽管目前对茎腐病的研究较多, 多个抗茎腐病QTL和基因都被定位, 但茎腐病的抗病机制仍不是很清楚。本研究利用不同抗性水平的近等基因系接种禾谷镰刀菌后进行转录组测序, 试图揭示玉米对禾谷镰刀菌的抗性机制, 对于研究寄主和病原菌互作, 加速茎腐病抗性育种进程具有重要意义。
1 材料与方法
1.1 玉米材料
早期研究中, 本实验室利用抗病自交系 1145和感病自交系Y331配制回交群体定位了2个抗茎腐病QTL, qRfg1和qRfg2, 抗病等位基因均来自1145。在高世代回交群体中, 分别挑选带有最短 qRfg1 (0.7 Mb)和qRfg2 (4 Mb)抗病区段的二个重组个体, 杂交后自交, 得到近等基因系NIL-R (qRfg1++/qRfg2++)和NIL-S (qRfg1--/qRfg2--)。利用GoldenGate 3KSNP (Illumina, San Diego, CA, USA)对2个NIL的基因组进行检测, 发现二者 99%以上的遗传背景和轮回亲本Y331一致。从2个近等基因系各取80粒种子进行发苗。种子经灭菌的ddH2O浸泡12 h后用70%的酒精消毒5 min。用灭菌的ddH2O冲洗一次, 再用10%的次氯酸钠(NaClO)浸泡30 min, 浸泡过程中每隔10 min摇晃一次, 再用灭菌的 ddH2O冲洗 3次后转置20%的多菌灵(carbendazol)溶液中浸泡 12 h, 用灭菌的 ddH2O冲洗干净后放入灭菌的发芽盒中。将发芽盒于16 h光照/8 h黑暗, 温度为(27±1)℃的光照培养箱中培养。待玉米根长到6~8 cm长时接种禾谷镰刀菌孢子液。
1.2 孢子液培养和接菌
利用绿豆培养基(mung bean medium)培养禾谷镰刀菌, 获取孢子液[25]。首先将禾谷镰刀菌在PDA培养基上扩繁, 待菌丝长满整个培养基时, 将整个PDA培养基接种到绿豆培养基中, 将绿豆培养基在180转 min-1的转速下暗培养7 d, 整个过程保持25℃的恒定温度。用纱布将绿豆培养基过滤, 去除菌丝、孢子囊和PDA培养基; 然后将过滤液在4℃离心机中以7500 × g的速度离心15 min后去除上清液;得到的孢子经灭菌的ddH2O悬浮混匀后, 浓度调至1×107个 mL-1, 于4℃冰箱保存备用。在转速为50×g的摇床上, 将玉米根在孢子液中浸泡 1 h, , 以保证孢子液悬浮; 再将接种后的玉米幼根转移至(27±1)℃, 16 h光照/8 h黑暗的恒温培养箱中培养。
1.3 总RNA提取
接种后0 h (将玉米幼根在孢子液中浸泡后立即取出)、6 h和18 h取样, 每个样品由10根玉米幼根组成, 设 2个生物学重复。立刻将样品用液氮冷冻并转移至-70℃保存备用。按照 TRIzol试剂盒(Invitrogen公司, 美国)说明书提取玉米幼根总RNA,并用RNeasy Mini试剂盒(Qiagen公司, 荷兰)处理。用Nanodrop分光光度计和1%琼脂糖凝胶检测所提取总RNA的浓度和质量。将所得的RNA样品用于转录组测序。
1.4 转录组测序
测序所用 RNA由贝瑞和康公司进行测序文库的构建并利用Illumina HiSeq 2000平台对12个样品的cDNA文库测序。测序所得的原始序列(raw reads)经过质量处理(去除短片段和低质量片段)后得到高质量clean reads。
1.5 转录组数据分析
利用Tophat 2.0.7将得到的clean reads与B73玉米基因组比对[26], 只保留能够特异比对到玉米基因组的片段用于后续分析。利用FPKM (Fragments Per Kilobase of exon model per Million mapped reads)计算基因的表达量, 随后用DEseq进行差异基因的筛选[27]。筛选条件为两基因之间的P-value/FDR < 0.05, Foldchange > 1。根据玉米基因组注释基因, 对于玉米中无功能注释信息的基因, 通过Blast2Go比对拟南芥数据库进行注释[28]。将所得差异基因进行Gene Ontology (GO)分析[29]。
1.6 荧光定量PCR
荧光定量PCR中用到的cDNA是利用M-MLV 第1链合成试剂盒(Invitrogen, 美国)合成。荧光定量PCR用到的染料为SYBR Green II (TaKaRa, 日本),仪器为Roter-Gene 6000 (Corbett Research, 悉尼, 澳大利亚)。PCR过程中以玉米GADPH作为内参基因,以2-ΔΔCt方法计算基因的相对表达量, 每个基因设2个生物学重复。荧光定量 PCR中用到的引物均用Primer 3.0设计, 所选基因及引物序列见表1。

表1 Real-time qRT-PCR分析中所用引物序列信息Table 1 Primers used in real-time qRT-PCR analysis
2 结果与分析
2.1 表型鉴定
在成株期和幼苗期, 分别评价 NIL-R和 NIL-S对禾谷镰刀菌的抗性。与NIL-S相比, NIL-R在2个时期均可显著提高玉米对禾谷镰刀菌的抗性(图1)。
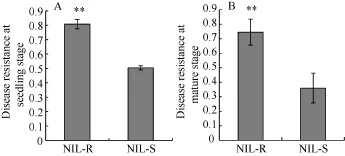
图1 玉米在成株期和幼苗期对禾谷镰刀菌的抗性Fig. 1 Disease resistance against F. graminearum at seedling and mature stages.
2.2 转录组测序质量及reads的比对
转录组测序后, 2个NIL共得到175 570 000个reads, 平均每个样品中reads数为29 261 667。对每个样品 reads进行质量检测, 各样品的 Q30均值为94.6, Q20均值为86.1。所有样品的2个生物学重复之间的皮尔森系数高于0.96。利用Tophat 2.0.7将所得reads与B73基因组参考序列比对, 共131 170 000 个 reads可比对到 B73基因组上, 平均每个样品有21 861 667个; 114 640 000个 reads可特异比对到B73基因组上, 平均每个样品有19 106 667个。
2.3 荧光定量PCR
在NIL-R和NIL-S中选取15个差异表达的基因,利用实时荧光定量PCR检测禾谷镰刀菌接种后0 h、6 h和18 h基因的表达情况。如图2, 两平台数据间的皮尔森相关系数为 0.78, 说明了 RNAseq数据的可靠性, 可进行禾谷镰刀菌接种后的转录组分析。
2.4 接种禾谷镰刀菌后 NIL-R和 NIL-S中基因的差异表达
接种禾谷镰刀菌后, 对NIL-R和NIL-S中基因的表达进行分析, 共检测到5583个基因差异表达(图3)。与对照组相比, 接种后6 h, 1495个基因在NIL-R中差异表达, 其中940个上调表达, 555个下调表达;NIL-S中有2964个基因差异表达, 其中1455个上调表达, 1509个下调表达。接种后18 h, NIL-R中有1846个基因差异表达, 其中 1317个上调表达, 529个下调表达; NIL-S中有 3856个基因差异表达, 其中2018个上调表达, 1838个下调表达。
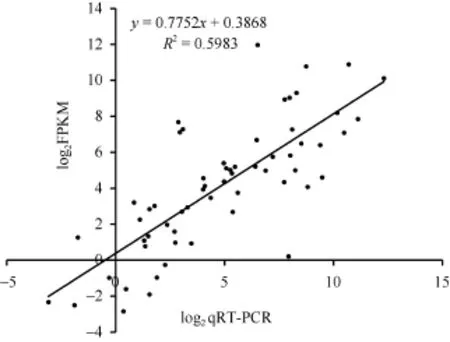
图2 Real-time qRT-PCR分析验证实验Fig. 2 Validation of DEGs by qRT-PCR

图3 接种禾谷镰刀菌后, NIL-R和NIL-S中差异表达的基因Fig. 3 The expression pattern of differentially expressed genes in NIL-R and NIL-S during infection of F. graminearum
接种后6 h, NIL-R和NIL-S中共有的差异基因1067个, NIL-R特有差异基因428个, NIL-S特有差异基因 1897个; 接种后 18 h, 共有差异表达基因1267个, NIL-R特有的579个, NIL-S特有的2589个(图4-A)。分析共有差异表达基因后发现, 接种后6 h,NIL-R和NIL-S有669个基因上调表达, 390个基因下调表达, 5个基因在NIL-R中上调而在NIL-S中下调表达, 3个基因在NIL-R中下调而在NIL-S中上调表达; 接种后18 h, NIL-R和NIL-S共有的差异表达基因中, 949个上调表达, 304个下调表达, 9个基因在NIL-R中上调而在NIL-S中下调表达, 5个基因在NIL-R中下调而在 NIL-S中上调表达。NIL-R和NIL-S共有差异表达基因中, 上调表达的基因多于下调表达的基因(图4-B)。
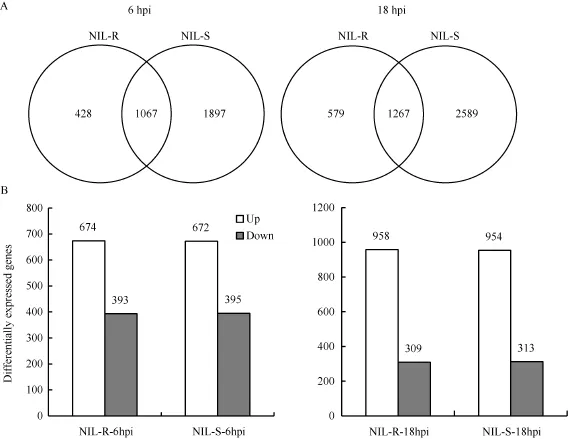
图4 接种禾谷镰刀菌后NIL-R和NIL-S中差异表达基因的维恩图Fig. 4 Veen diagrams of all the differentially expressed genes (DEGs) in the resistant NIL-R and susceptible NIL-S after inoculation of F. graminearum
2.5 NIL-R特异上调表达的抗病相关基因
接种后6 h, NIL-R中特有的428个差异基因中,上调表达的266个, 下调表达的162个; 接种后18 h,NIL-R中特有的579个差异基因中, 上调表达的359个, 下调表达的220个。为研究NIL-R对禾谷镰刀菌的抗性响应, 对NIL-R中2个时间点特有上调表达的基因进行分析。接种后6 h, NIL-R中多种和抗病相关的基因特有上调表达(表2), 包括编码ET合成酶(1-aminocyclopropane-1-carboxylate oxidase 1,ACO)和乙烯信号途径中 ethylene-responsive factorlike protein 1 (ERF1)、EIN3-binding F-box protein 1 (EBF1)、ETHYLENE-INSENSITIVE3-like 1 (EIL1),病程相关蛋白 PR4、索马甜蛋白(putative thaumatin domain family protein)、germin-like protein 2 fragment (GLP2 fragment)、脂转运蛋白(lipid-transfer protein, LTP)及其他抗性相关蛋白GST23、过氧化物酶(peroxidase 16, POD16)、LRR激酶(leucine-rich repeat protein kinase family protein)的基因。
接种后18 h, NIL-R中有359个基因特有上调表达, 其中包括编码解毒蛋白 ABC类转运蛋白(ABC-2 type transporter family protein)、重金属转运蛋白(MATE efflux family protein), 及编码病程相关蛋白(PR1、PR4), 编码激酶类基因和苯丙烷代谢途径中合成木质素的羟基肉桂酰辅酶A奎尼羟基肉桂转移酶(hydroxycinnamoyl-CoA hikimate/quinate hydroxycinnamoyl transferase, HCT)的基因等(表3)。
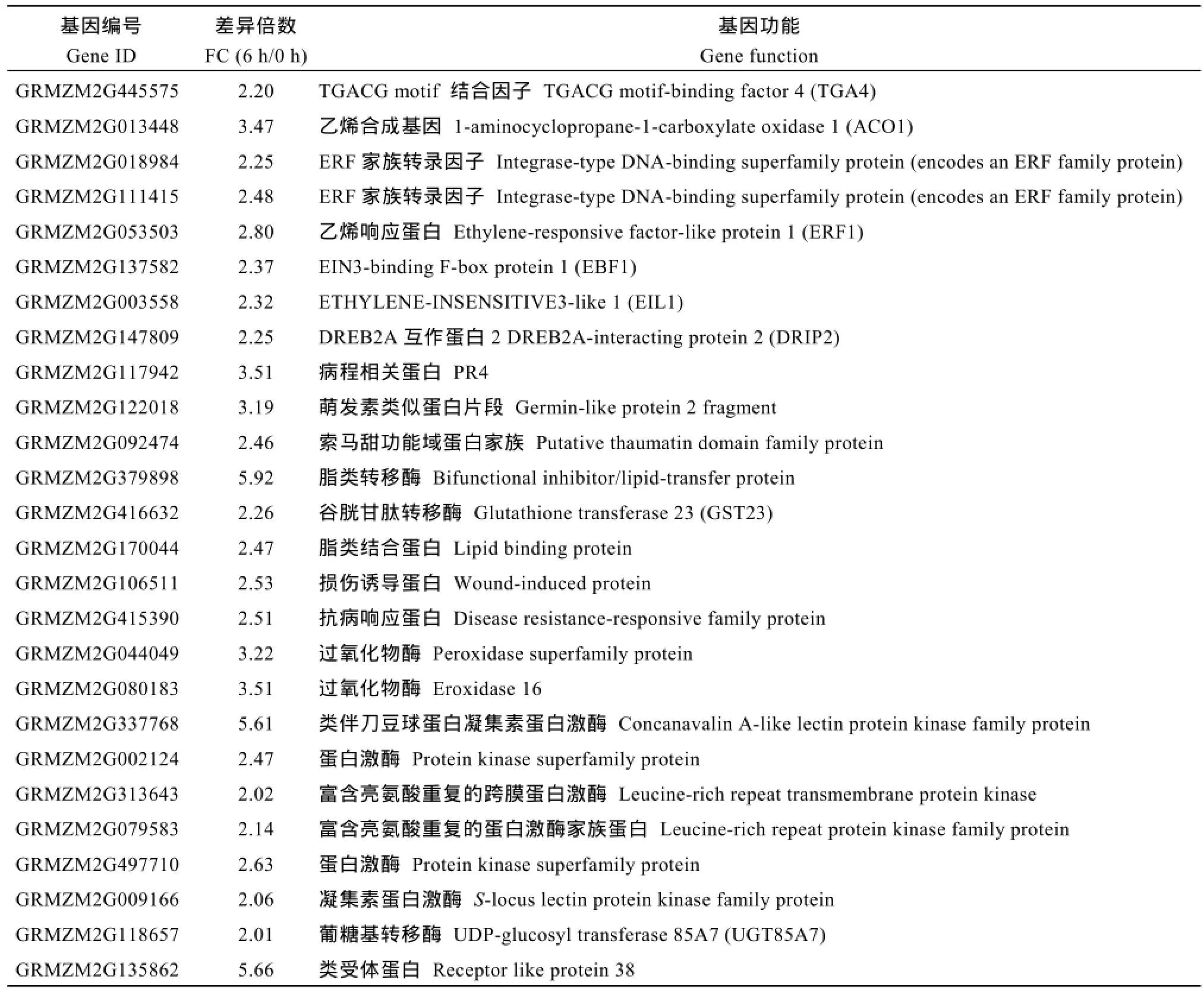
表2 接种后6 h, NIL-R中特异上调表达的抗病相关基因Table 2 Disease resistance genes specifically up-regulated in NIL-R at 6 hai

表3 接种后18 h, NIL-R中特异上调表达的抗病相关基因Table 3 Disease resistance genes specifically up-regulated in NIL-R at 18 hai
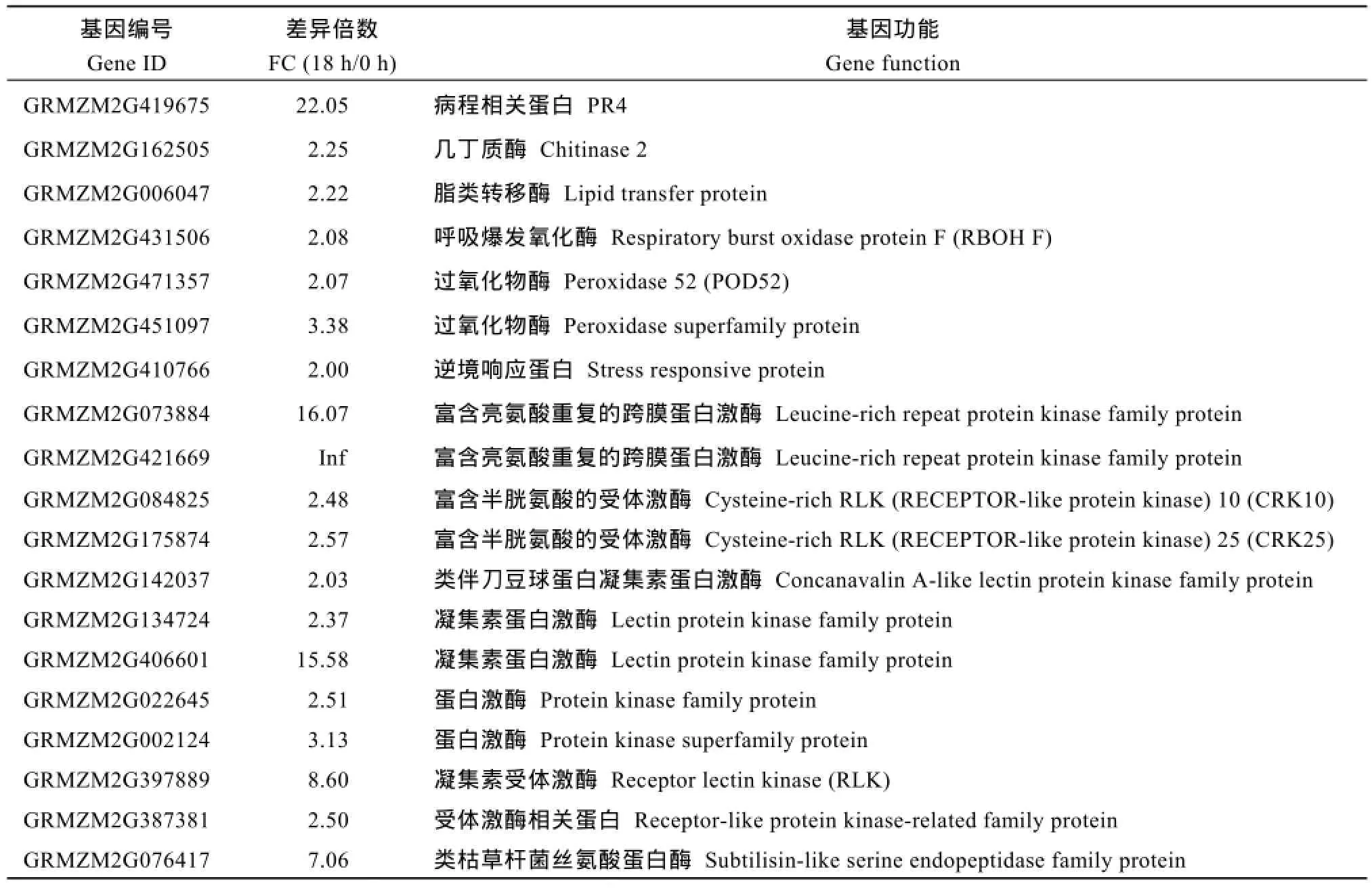
(续表3)
接种后 6 h, 5个基因(GRMZM2G429000、GRMZM5G805132、GRMZM2G456217、GRMZM5 G847744、GRMZM2G576460)在NIL-R中上调而在NIL-S中下调表达。其中 GRMZM2G429000和GRMZM5G805132编码赤霉素响应蛋白 2 (Gibberellin responsive 2, GAR2)。
接种后 18 h, 9个基因(GRMZM5G884819、GRMZM2G084583、GRMZM2G392148、GRMZM2 G155216、GRMZM2G353753、AC207722.2_FG009、AC190623.3_FG001、GRMZM2G081239、GRMZM2 G092427)在NIL-R中上调而在NIL-S中下调表达。其中 GRMZM2G084583编码 Typical P-type R2R3 Myb protein fragment, GRMZM2G081239 编码NOD26-like membrane integral protein, 5个基因编码光吸收有关蛋白, 2个为功能未知蛋白。
2.6 NIL-R和NIL-S间差异表达的基因
接种后0 h, 2958个基因在NIL-R和NIL-S间差异表达, 其中 1170个基因在 NIL-R中表达量较高,1788个基因在NIL-S中表达量较高(图5)。接种后6 h, 431个基因在NIL-R和NIL-S间差异表达, 其中83个基因在 NIL-R中表达量较高, 348个基因在NIL-S中表达量较高。接种后18 h时, 1292个基因在NIL-R和NIL-S间差异表达, 其中291个基因在NIL-R中表达量较高, 1001个基因在NIL-S中表达量较高(图5)。两近等基因系之间差异表达的基因在0 h最多, 接种后6 h最少。

图5 NIL-R和NIL-S间差异表达的基因Fig. 5 Differentially expressed genes between NIL-R and NIL-S
2.7 NIL-R 通过组成型高表达抗性相关基因抵御禾谷镰刀菌入侵
对NIL-R和NIL-S在接种后0 h的差异表达基因进行分析, 获得二者间的本底差异。对 NIL-R中表达量较高的1170个基因进行GO分析, 功能归为3类, 即生物学过程(biological process)、分子功能(molecular function)和细胞组分(cellular component)。就生物学过程中富集的go term而言, 和抗病有关的go term在1170个基因中显著富集(FDR < 0.05)。植物激素JA、SA和ET相关的生物学过程的基因显著富集, 这些基因参与JA合成、响应和信号介导途径,ET合成、响应过程, SA合成、响应和信号介导途径以及SA介导的系统获得性抗性(SAR)。JA合成途径中编码12-氧-植物二烯酸还原酶(12-oxophytodienoic acid reductase, OPR3)和脂氧化酶(lipooxygenase, LOX)的基因, JA 信号途径中的JAZ1、JAZ3、JAZ10等基因在NIL-R中表达量较高。乙烯合成酶, 如ACC、ACO31、ACS2 (Acc synthase 2)、ACS7, 乙烯信号途径基因, 如2个ERF-1 (Ethylene responsive element binding factor 1)、1个ERF4 在 NIL-R中高表达。此外, 次生代谢有关的生物学过程, 如苯丙烷代谢途径、肉桂酸、香豆酸、植物抗毒素合成途径在NIL-R本底表达量较高的基因中显著富集(图6)。编码苯丙烷代谢途径的关键酶苯丙氨酸解氨酶(PHE ammonia lyase 1, PAL1)、PAL2的基因, 编码肉桂酸-乙醇脱氢酶(cinnamyl-alcohol dehydrogenase, CAD)、木质素合成酶HCT的基因在NIL-R中表达量较高。由此表明, NIL-R可以通过抗病相关基因的组成型高表达, 预先储存大量的抗病次生代谢产物来抵抗病原菌的入侵。

图6 NIL-R对照组中表达量较高基因的GO功能分析Fig. 6 GO analysis of genes more abundant in NIL-R control samples
2.8 禾谷镰刀菌侵染后 NIL-R与 NIL-S之间差异表达的抗性相关基因
NIL-R可以通过抗病相关基因的组成型高表达,阻碍病原菌入侵来提高抗性。此外, 抗、感近等基因系在接种6 h和18 h后有大量差异表达的抗性相关基因。接种后6 h, 在NIL-R中表达量较高的抗病相关基因主要编码类黄酮醇合成酶(flavonol synthase-like protein)、transmembrane BAX inhibitor motif-containing protein 4 (TMBIM 4)、LTP、germin-like protein (GLP) subfamily 1 member 11、过氧化物酶(peroxidase R15, POD R15)、LRR蛋白激酶(leucine-rich repeat protein kinase family protein)、CDPK 相关激酶(CDPK-related kinase 1, CRK1;CDPK-related kinase 3, CRK3)等蛋白的基因(表4)。

表4 接种后6 h, NIL-R中高表达的抗病相关基因Table 4 Disease resistance genes more abundant in NIL-R at 6 hai
接种18 h后, 在NIL-R中表达量较高的抗病相关基因包括ET合成相关基因ACO、ACO1, JA合成基因 LOX10, 编码多种激酶, 如 CRK1、BAK1 (BRI1-associated receptor kinase 1)、富含半胱氨酸受体激酶 10 (cysteine-rich RECEPTOR-like protein kinase 10, CRK10)、LRR激酶、细胞壁相关激酶(wall-associated kinase 2, WAK2)、LRR受体激酶(LRR-RLK family protein)的基因, 编码木质素合成关键酶HCT、解毒蛋白PDR1、多药抗性相关蛋白2 (multidrug resistance associated protein 2, MDR2)、POD R15的基因等(表5)。
3 讨论
禾谷镰刀菌是一类危害严重的病原菌, 它可以侵染大麦、小麦、玉米等多种禾谷类作物。禾谷镰刀菌属半活体营养型病原菌, 其特点为病原菌侵入寄主后先以活体营养型存活, 当寄主出现局部组织坏死和菌丝大量繁殖后, 病原菌再以坏死营养型在寄主组织内繁殖。SA、JA/ET在植物与病原菌的互作中发挥重要作用; SA主要介导对活体营养型和半活体营养型病原菌的抗性; JA/ET主要针对坏死营养型病原菌的入侵[30]。
JA可正向调节由禾谷镰刀菌引起的小麦赤霉病的抗性[16]。Xiao等指出, JA合成酶AOS和OPR3在抗、感赤霉病小麦中都被诱导, 而JA信号途径中的COI1和JAZ只在抗病材料中被诱导, 因此JA可正向调控小麦对F. graminearum的抗性[31]。ET合成基因只在感病小麦中被诱导, 抗性小麦可通过抑制ET信号途径而提高抗性, 故 ET可负向调控小麦对 F. graminearum的抗性[31]。而有报道指出, ET合成基因ACO在抗性材料中被诱导, 故ET也可正向调控小麦对 F. graminearum 的抗性[32-33]。本研究中,JA/ET合成和信号途径相关基因在接种后0 h、18 h 的NIL-R中表达水平较高; 且ET合成基因ACO, ET信号途径中的基因ERF、EBF1、EIL1在接种后6 h 的NIL-R中特异上调表达, 说明本研究中JA/ET合成和信号途径可正向调控玉米对F. graminearum的抗性。
次生代谢产物在植物的生物和非生物胁迫中发挥重要作用。苯丙烷代谢途径可产生木质素、黄酮类、植物抗毒素等多种次生代谢产物[34]。PAL是苯丙烷代谢途径的关键酶, 可以催化苯丙氨酸向肉桂酸的转化, 肉桂酸经 C4H转化为香豆酸, 随后经4CL转化为4-酰基-辅酶A。4-酰基-辅酶A可经CAD 和HCT转化为木质素, 也可经CHS和FLS转化为黄酮醇。多个报道指出, 镰刀属病原菌侵染可诱导PAL的表达[19,22,24]。用 F. verticillioides接种不同穗腐病抗性玉米发现, 与感病材料相比, 3个PAL基因在抗性材料中被较高水平的诱导, 2个PAL在抗性材料中本底表达水平较高; 此外木质素合成相关基因Caffeoyl-o-methyltransferase 1 (COMT1)在未接种的抗性材料中表达量较高[24]。Ye等[35]研究表明, 接种后 6 h, 抗病近等基因系玉米幼根表皮侵入的 F. graminearum菌丝少于感病近等基因系, 此结果表明与感病近等基因系相比, 抗性材料可抑制病原菌入侵。苯丙烷代谢途径产物酚类在抗茎腐病玉米近等基因系[35]和抗赤霉病小麦材料[36]中含量较高, 而酚类物质可与细胞壁共价结合, 致使病原菌侵染位置细胞壁坏死而抵抗病原菌入侵; 此外, 木质素积累可使玉米根部外皮组织加厚, 进一步抑制病原菌的入侵[35]。接种F. culmorum后, 2个抗病小麦品种中木质素积累量较感病小麦品种中高[22]。本研究的NIL-R中高表达的基因在苯丙烷合成途径中显著富集; 此外还包括肉桂酸、香豆酸、植物抗毒素等次生代谢产物合成途径。多个苯丙烷代谢途径的关键基因, 如PAL1、PAL2、CAD、HCT等在接种后0 h 的NIL-R中表达量较高。结果表明, 苯丙烷代谢途径可通过组成型抗性提高玉米对禾谷镰刀菌的抗性。

表5 接种后18 h, NIL-R中高表达的抗病相关基因Table 5 Disease resistance genes more abundant in NIL-R at 18 hai
禾谷镰刀菌可分泌DON, 该毒素可作为有毒因子参与到病原菌侵染寄主的过程中[10]。DON并非病原菌生长必需, 但有报道称毒素影响病原菌致病性。与野生型相比, 毒性合成基因tri-5突变株在侵染小麦时致病力减弱[12-13]。研究报道, 动物中DON可与60S核糖体亚基结合来抑制蛋白质的合成进而激活细胞衰亡程序导致细胞死亡[8-10]。DON可体外抑制小麦核糖体中蛋白的合成[11]。所以有研究认为,DON可通过抑制寄主体内抗性蛋白的合成来延缓或抑制寄主的抗性。此外, DON不仅可引起细胞内过氧化氢的积累引起细胞程序化死亡, 还可以激活抗性响应基因病程相关蛋白的表达[15]。寄主细胞可通过诱导毒素降解蛋白的合成降解毒素或毒素转运蛋白的合成将毒素转运出胞质[15]。接种后 18 h, 编码ABC-transport family protein、Heavy metal transport protein、MATE efflux family protein的基因在NIL-R中特异上调表达。PDR1、MRD2在接种后18 h的NIL-R中表达量较高。故NIL-R可通过诱导毒素降解或转运基因的表达而降低毒素对寄主细胞的毒性, 抑制F. graminearum的进一步入侵。
RLK是一类位于质膜上的受体激酶, 它可被病原菌的相关分子模式PAMP或MAMP识别, 而激活下游的PTI抗性反应。FLS2是一类位于质膜的LRR型受体激酶, 该激酶可识别细菌的鞭毛蛋白而提供对细菌的抗性; BAK1是一类LRR型受体激酶, 它可通过调节BRI1参与到FLS2调节的PTI反应中[37]。本研究中多种RLK基因在接种后18 h的NIL-R中特异上调表达。BAK1基因在接种后18 h的NIL-R中表达量较高。说明 PTI免疫反应在玉米抵抗禾谷镰刀菌侵染的过程中发挥重要作用。
4 结论
与NIL-S相比, NIL-R可能主要依赖本底高表达抗性相关基因(如JA/ET合成和信号途径相关基因、苯丙烷代谢途径合成基因)来抵抗病原菌的入侵; 病原菌入侵后, 编码病程相关蛋白(PR)、木质素合成、DON降解或转运等基因的高表达限制病原菌的进一步扩展和繁殖; 此外, 编码受体激酶的基因在NIL-R中的高表达说明了 PTI免疫反应在玉米抵抗禾谷镰刀菌侵染的过程中也发挥了重要作用。
References
[1] Yang Q, Yin G M, Guo Y L, Zhang D F, Chen S J, Xu M L. A major QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet, 2010, 121: 673-687
[2] Ali M L, Taylor J H, Jie L, Sun G L, William M, Kasha K J, Reid L M, Pauls K P. Molecular mapping of QTLs for resistance to Gibberella ear rot, in corn, caused by Fusarium graminearum. Genome, 2005, 48: 521-533
[3] Schweiger W, Steiner B, Ametz C, Siegwart G, Wiesenberger G,Berthiller F, Lemmens M, Jia H Y, Adam G, Muehlbauer G J. Transcriptomic characterization of two major Fusarium resistance quantitative trait loci (QTLs), Fhb1 and Qfhs. Ifa-5A,identifies novel candidate genes. Mol Plant Pathol, 2013, 14:772-785
[4] Boddu J, Cho S, Kruger W M, Muehlbauer G J. Transcriptome analysis of the barley-Fusarium graminearum interaction. Mol Plant-Microbe Interact, 2006, 19: 407-417
[5] Goswami R S, Kistler H C. Pathogenicity and in planta mycotoxin accumulation among members of the Fusarium graminearum species complex on wheat and rice. Phytopathology, 2005, 95: 1397-1404
[6] Urban M, Daniels S, Mott E, Hammond-Kosack K. Arabidopsis is susceptible to the cereal ear blight fungal pathogens Fusarium graminearum and Fusarium culmorum. Plant J, 2002, 32:961-973
[7] McMullen M, Jones R, Gallenberg D. Scab of wheat and barley:a re-emerging disease of devastating impact. Plant Dis, 1997, 81:1340-1348
[8] Rocha O, Ansari K, Doohan F M. Effects of trichothecene mycotoxins on eukaryotic cells: a review. Food Addit Contam,2005, 22: 369-378
[9] Pestka J J, Zhou H R, Moon Y, Chung Y J. Cellular and molecular mechanisms for immune modulation by deoxynivalenol and other trichothecenes: unraveling a paradox. Toxicol Lett, 2004, 153:61-73
[10] Pestka J J. Deoxynivalenol-induced proinflammatory gene expression: Mechanisms and pathological sequelae. Toxins, 2010,2: 1300-1317
[11] Miller J D, Ewen M A. Toxic effects of deoxynivalenol on ribosomes and tissues of the spring wheat cultivars Frontana and Casavant. Nat Toxins, 1997, 5: 234-237
[12] Desjardins A E, Proctor R H, Bai G H, McCormick S P, Shaner G,Buechley G, Hohn T M. Reduced virulence of trichothecenenonproducing mutants of Gibberella zeae in wheat field tests. Mol Plant-Microbe Interact, 1996, 9: 775-781
[13] Langevin F, Eudes F, Comeau A. Effect of trichothecenes produced by Fusarium graminearum during Fusarium head blight development in six cereal species. Eur J Plant Pathol,2004, 110: 735-746
[14] Harris L, Desjardins A E, Plattner R, Nicholson P, Butler G,Young J, Weston G, Proctor R, Hohn T. Possible role of trichothecene mycotoxins in virulence of Fusarium graminearum on maize. Plant Dis, 1999, 83: 954-960
[15] Desmond O J, Manners J M, Stephens A E, Maclean D J, Schenk P M, Gardiner D M, Munn A M, Kazan K. The Fusarium mycotoxin deoxynivalenol elicits hydrogen peroxide production,programmed cell death and defence responses in wheat. Mol Plant Pathol, 2008, 9: 435-445
[16] Jia H Y, Cho S, Muehlbauer G J. Transcriptome analysis of a wheat near-isogenic line pair carrying Fusarium head blightresistant and -susceptible alleles. Mol Plant-Microbe Interact,2009, 22: 1366-1378
[17] Poppenberger B, Berthiller F, Lucyshyn D, Sieberer T,Schuhmacher R, Krska R, Kuchler K, Glossl J, Luschnig C,Adam G. Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J Biol Chem, 2003, 278: 47905-47914
[18] Muhitch M J, McCormick S P, Alexander N J, Hohn T M. Transgenic expression of the TRI101 or PDR5 gene increases resistance of tobacco to the phytotoxic effects of the trichothecene 4,15-diacetoxyscirpenol. Plant Sci, 2000, 157:201-207
[19] Boddu J, Cho S, Muehlbauer G J. Transcriptome analysis of trichothecene-induced gene expression in barley. Mol Plant-Microbe Interact, 2007, 20: 1364-1375
[20] Gardiner S A, Boddu J, Berthiller F, Hametner C, Stupar R M,Adam G, Muehlbauer G J. Transcriptome analysis of the barleydeoxynivalenol interaction: evidence for a role of glutathione in deoxynivalenol detoxification. Mol Plant-Microbe Interact, 2010,23: 962-976
[21] Schweiger W, Boddu J, Shin S, Poppenberger B, Berthiller F,Lemmens M, Muehlbauer G J, Adam G. Validation of a candidate deoxynivalenol-inactivating UDP-glucosyltransferase from barley by heterologous expression in yeast. Mol Plant-Microbe Interact, 2010, 23: 977-986
[22] Kruger W M, Pritsch C, Chao S, Muehlbauer G J. Functional and comparative bioinformatic analysis of expressed genes from wheat spikes infected with Fusarium graminearum. MolPlant-Microbe Interact, 2002, 15: 445-455
[23] Zhu Q H, Stephen S, Kazan K, Jin G, Fan L, Taylor J, Dennis E S,Helliwell C A, Wang M B. Characterization of the defense transcriptome responsive to Fusarium oxysporum-infection in Arabidopsis using RNA-seq. Gene, 2013, 512: 259-266
[24] Lanubile A, Ferrarini A, Maschietto V, Delledonne M, Marocco A,Bellin D. Functional genomic analysis of constitutive and inducible defense responses to Fusarium verticillioides infection in maize genotypes with contrasting ear rot resistance. BMC Genomics, 2014, 15: 710
[25] Buerstmayr H, Steiner B, Lemmens M, Ruckenbauer P. Resistance to Fusarium head blight in winter wheat: heritability and trait associations. Crop Sci, 2000, 40: 1012-1018
[26] Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley D R,Pimentel H, Salzberg S L, Rinn J L, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols, 2012, 7: 562-578
[27] Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol, 2010, 11: R106
[28] Conesa A, Götz S, García-Gómez J M, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 2005,21: 3674-3676
[29] Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry J M, Davis A P, Dolinski K, Dwight S S, Eppig J T. Gene Ontology: tool for the unification of biology. Nat Genet, 2000, 25:25-29
[30] Gimenez-Ibanez S, Solano R. Nuclear jasmonate and salicylate signaling and crosstalk in defense against pathogens. Front Plant Sci, 2013, 4: 7
[31] Xiao J, Jin X H, Jia X P, Wang H Y, Cao A Z, Zhao W P, Pei H Y,Xue Z K, He L Q, Chen Q G, Wang X. Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genomics, 2013, 14: 197
[32] Li G L, Yen Y. Jasmonate and ethylene signaling pathway may mediate Fusarium head blight resistance in wheat. Crop Sci, 2008,48: 1888-1896
[33] Ding L N, Xu H B, Yi H Y, Yang L M, Kong Z X, Zhang L X,Xue S L, Jia H Y, Ma Z Q. Resistance to hemi-biotrophic F. graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PloS One, 2011, 6: e19008
[34] Steiner B, Kurz H, Lemmens M, Buerstmayr H. Differential gene expression of related wheat lines with contrasting levels of head blight resistance after Fusarium graminearum inoculation. Theor Appl Genet, 2009, 118: 753-764
[35] Ye J R, Guo Y L, Zhang D F, Zhang N, Wang C, Xu M L. Cytological and molecular characterization of quantitative trait locus qRfg1, which confers resistance to Gibberella stalk rot in maize. Mol Plant-Microbe Interact, 2013, 26: 1417-1428
[36] Hamzehzarghani H, Kushalappa A C, Dion Y, Rioux S, Comeau A, Yaylayan V, Marshall W D, Mather D E. Metabolic profiling and factor analysis to discriminate quantitative resistance in wheat cultivars against Fusarium head blight. Physiol Mol Plant Pathol, 2005, 66: 119-133
[37] Chinchilla D, Zipfel C, Robatzek S, Kemmerling B,Nürnberger T, Jones J D, Felix G, Boller T. A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature, 2007, 448: 497-500
Transcriptional Analysis of Maize Resistance against Fusarium graminearum
LIU Yong-Jie, MA Chuan-Yu, MA Xue-Na, and XU Ming-Liang*
National Maize Improvement Center of China, China Agricultural University, Beijing 100193, China
Gibberella stalk rot, caused by Fusarium graminearum (teleomorph, Gibberella zeae), is one of the most devastating soil-borne diseases in maize. It seriously decreases maize yield and quality. Molecular mapping led to the identification of two QTLs, qRfg1 and qRfg2, on chromosomes 10 and 1 respectively, conferring resistance to Gibberella stalk rot. In order to characterize the defense mechanism of maize against F. graminearum, NIL-R with resistant alleles at both QTLs and NIL-S with the susceptible alleles at both QTLs were generated and used in transcriptome analysis. After inoculation of young seedling roots of both NILs with the F. graminearum spores, the inoculated roots were sampled at 0, 6, and 18 hours after inoculation (hai) for transcriptome analysis using RNAseq. The basal difference was achieved by the comparison between control samples. In total,2958 genes were differentially expressed between control samples of NIL-R and NIL-S, among which 1170 genes were more abundant in NIL-R. GO analysis revealed that genes involved in biological processes related to JA/ET and SA biosynthesis, JA/ET mediated signaling pathway and SA mediated signaling pathway were significantly enriched. Phenylpropanoid biosynthesis process was enriched in the genes more abundant in NIL-R and genes encoding enzymes involved in phenylpropanoid biosynthesis like PAL, 4CL2, CAD, and HCT were more abundant in NIL-R. There were 431 genes differentially expressed between NIL-R and NIL-S at 6 hai, among which 83 genes were more abundant in NIL-R. Genes encoding pathogenesis-related (PR) proteins like lipid-transfer protein and germin-like protein were more abundant in NIL-R. Among the 1292 genes differentially expressed between NIL-R and NIL-S. At 18 hai, 291 genes were more abundant in NIL-R. Genes involved in ET biosynthesis like ACO and JA biosynthesis like LOX were more abundant in NIL-R. Genes involved in DON detoxification like PDR1 and MDR2 were more abundant in NIL-R. After inoculation with F. graminearum, 428 genes were exclusively up-regulated in NIL-R at 6 hai compared with control. Genes involved in ET biosynthesis and ET-mediated signaling pathway like ACO, ERF, EBF1, and EIL1 and pathogenesis-related genes like PR1, OSM34, and germin-like protein were exclusively up-regulated in NIL-R. At 18 hai, 359 genes were exclusively up-regulated in NIL-R compared with control. Pathogenesis-related genes like PR1, PR4, and genes encodingthe transporters of DON out of cytoplasm like ABC transport family protein, heavy metal transport protein and MATE efflux family protein were exclusively up-regulated in NIL-R. All these results indicate that NIL-R can increase the resistance of maize to F. graminearum by the constitutive resistance characterized by the higher expression of genes related to defense responses. Genes involved in defense responses exclusively up-regulated in NIL-R and higher expression level of disease resistance genes in NIL-R at 6 and 18 hai may restrict the pathogen invasion after infection. The phenylpropanoid biosynthesis pathway and DON-detoxification proteins identified in this study are important for the resistance against F. graminearum infection.
Maize; Stalk rot; Transcriptome; Resistance; JA/ET; Phenylpropanoid
10.3724/SP.J.1006.2016.01122
本研究由引进国际先进农业科学技术计划(948计划)项目(2003-Q04)资助。
This study was supported by the Program of Introducing International Super Agricultural Science and Technology (948 Program) (2011-G15).*
(Corresponding author): 徐明良, E-mail: mxu@cau.edu.cn, Tel: 010-62733166
联系方式: E-mail: liu_yongj@126.com
Received(
): 2016-01-11; Accepted(接受日期): 2016-05-09; Published online(网络出版日期): 2016-05-30.
URL: http://www.cnki.net/kcms/detail/11.1809.S.20160530.0905.008.html
