[Cl-M-PH3]2(M=Cu,Ag,Au)复合物亲金属效应的理论计算
2016-08-27王一波
孙 涛,王一波▲
(1贵州大学 网络与信息化管理中心,贵州 贵阳 550025;2贵州省高性能计算化学重点实验室, 贵州 贵阳 550025)
[Cl-M-PH3]2(M=Cu,Ag,Au)复合物亲金属效应的理论计算
孙涛1,2,王一波1,2▲
(1贵州大学 网络与信息化管理中心,贵州贵阳550025;2贵州省高性能计算化学重点实验室, 贵州贵阳550025)
依照高级电子相关校正理论CCSD(T)/ aug-cc-pVTZ的水平,对[Cl-M-PH3]2(M=Cu,Ag,Au)复合物的亲金属相互作用进行了研究,发现相互作用能整体差别不大,亲银作用较强,亲金次之,亲铜较弱。用对称性匹配微扰理论(SAPT)对这些复合物的结合能进行能量分解分析,结果表明由Cu到Au,静电能、诱导能和色散能逐渐增大,诱导占主要吸引作用的58 %以上,起主导作用,静电和色散能相差不大,其中[Cl-Ag- PH3]2的诱导能比[Cl-Au- PH3]2大。
亲金属效应,分子间相互作用,SAPT,能量分解
0 引言
近几十年来,对IB族金属簇合物及金属线分子的研究,发现在许多这类分子中金属原子之间存在相互作用。按常规,在分子中非键而带同类电荷的原子应互相推斥,但实际上许多IB族化合物分子中非键金属原子之间存在相互吸引作用,我们称之为IB族的亲金属效应[1-5]。Pykkö等人对IB族亲金属相互作用进行广泛的理论研究,分别使用HF和MP2方法计算[X-Au-PH3]2(X=H,CH3,SCH3)的亲金相互作用,为了降低两单体之间的偶极—偶极相互作用,将P-Au-Au-P二面角固定为90°。计算结果表明在HF水平两单体之间是相互排斥的而在MP2水平是相互吸引的,因此Pykkö认为亲金相互作用来源于电子相关,其中相对论效应增强了这种亲金属效应[6-7]。1997年,Pykkö在MP2水平对[Cl-Au-PH3]2中的Au原子分别使用准相对论和非相对论赝势基组,结果表明相对论效应重要但是并不占主导作用[8]。
Magnko等人使用LMP2方法,对[X-M-PH3]2(X=H,Cl;M=Cu,Ag,Au)计算结果表明,有Cu到Au,亲金属效应逐渐增强;电子相关不是纯粹的吸引作用,分子内的电子相关是排斥的;并对分子间的电子相关能进行能量分解,结果表明亲金属相互作用主要来自于色散力和离子激发[9-10]。O’Grady等人使用MP2、DFT和CCSD(T)方法,对[X-M-PH3]2(M= Cu,Ag,Au ,[111])计算再次证明在MP2水平,有Cu到Au亲金属相互作用逐渐增强,与Pykkö和Magnko等人得到的计算结果相一致[8,10];但是在CCSD(T)水平为[Cl-Ag-PH3]2的亲银相互作用最大,其次为[Cl-Au-PH3]2和[Cl-Cu-PH3]2,[Cl-[111]-PH3]2最小,得到与MP2不一致的结果[11]。
在过去的十几年,IB族亲金属效应被广泛研究,就目前所知道的,先前的主要工作是使用MP2、LMP2、DFT、CCSD(T)等方法计算亲金属效应,Pykkö等人曾指出亲金属效应有待深入研究[8];本文在CCSD(T)高级电子相关计算水平研究[Cl-M-PH3]2(M=Cu,Ag,Au)复合物的亲金属效应,并使用对称性匹配微扰理论(Symmetry-Adapted Perturbation Theory,SAPT)对其进行能量分解,研究亲金属相互作用的本质。
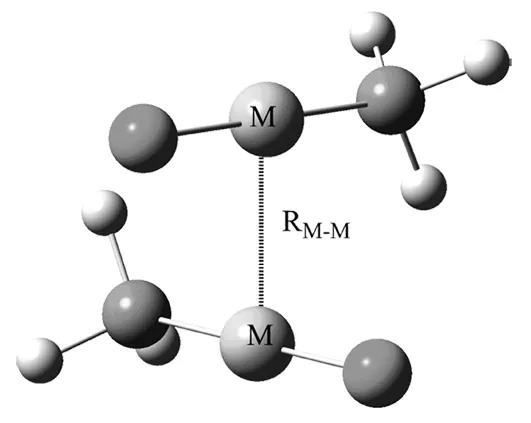
图1 [Cl-M-PH3]2(M=Cu,Ag,Au)的结构模型Fig.1 The structure of [Cl-M-PH3]2(M=Cu,Ag,Au)model
1 计算方法及过程
单体几何结构优化采用Gaussian09[18]程序,其余计算全部用Molpro2012.1[19]程序完成。
2 结果与讨论
2.1Cl-M-PH3(M=Cu,Ag,Au)
Cl-M-PH3单体具有C3v对称性,几何结构优化数据见表 1。Cl-Au-PH3的r(M-Cl)和r(M-P)计算结果与实验值非常接近,由此也可以看出MP2电子相关校正方法对单体的几何结构优化是正确的。三种单体的r(P-H)和∠H-P-M差别很小。从Cu 到Ag 的r(M-Cl)和r(M-P)显著增长(0.2Å左右);从Ag到Au 的r(M-Cl)变化不大,而r(M-P)明显减小(0.07Å)。Bowmaker等人认为是相对论效应使Au原子轨道收缩和相对论效应加强了Au的d轨道与PH3基团形成反馈π键,因此r(Au-P)键长减小[20]。
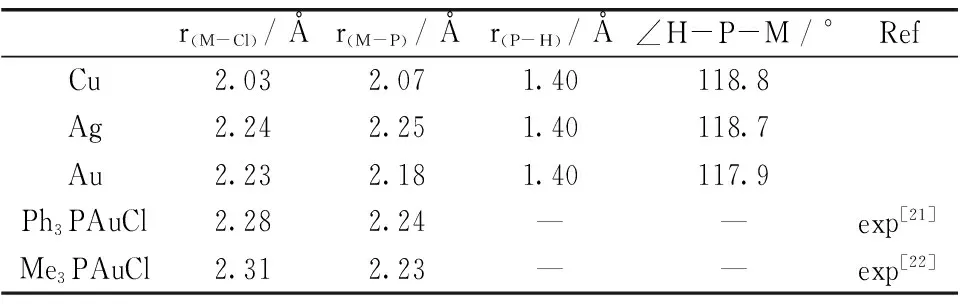
表1 Cl-M-PH3(M=Cu,Ag,Au)单体优化几何结构参数Tab.1 Optomised geometric parameters for Cl-M-PH3(M=Cu,Ag,Au)
2.2[Cl-M-PH3]2(M=Cu,Ag,Au)
为了降低两单体之间的偶极—偶极相互作用,将P-M-M-P二面角固定为90°,逐渐改变分子间距离RM-M,分别使用MP2和CCSD(T)方法计算其相互作用能。其平衡距离及相互作用能见表2。
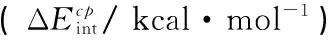
Tab.2Calculated equilibrium distances [Re / Å] and

CuAgAuReΔEintΔEcpintBSSEReΔEintΔEcpintBSSEReΔEintΔEcpintBSSEMP22.96-5.93-4.691.242.98-8.13-7.150.983.078.71-8.160.55CCSD(T)2.96-6.02-4.691.323.10-6.40-5.281.123.345.97-4.841.13
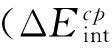
Cu、Ag和Au的范德华半径分别为1.43Å、1.70Å和1.66Å,CCSD(T)计算结果显示除了[Cl-Ag-PH3]2分子间距离Re(Ag-Ag)小于其范德华半径之和外,[Cl-M-PH3]2(M=Cu,Au)分子之间的距离Req(M-M)并不比其范德华半径和小。由于目前还未有实验上的[Cl-M-PH3]2(M=Cu,Ag,Au)数据结果支持,与其类似的化合物(Me3P)AuCl的晶体显示呈螺旋型的多聚链,Au(I)-Au(I)间的平均距离为3.34 Å[22],这与CCSD(T)计算的[Cl-Au-PH3]2分子之间的距离Re(Au-Au)结果相一致。
Pykkö等人认为亲金相互作用来源于电子相关,Magnko的计算结果表明在亲金属效应中色散力起主要作用。为了分析亲金属效应的本质作用,使用SAPT对[Cl-M-PH3]2(M=Cu,Ag,Au)进行能量分解。结果见表3。
Patkowski等人使用SAPT对闭壳层金属双原子二聚体相互作用计算结果并不理想[25],由表2 和表3可以看出SAPT与CCSD(T)计算结果非常接近,除了[Cl-Au-PH3]2误差较大10.54 %外,[Cl-Cu-PH3]2和[Cl-Ag-PH3]2误差均在3 %以内。综上所述,SAPT对[Cl-M-PH3]2(M=Cu,Ag,Au)的计算是准确的。虽然SAPT计算结果显示由Cu到Au的相互作用能逐渐增大,与CCSD(T)结果不完全一致,但是在这里并不影响我们对能量成份的分析。
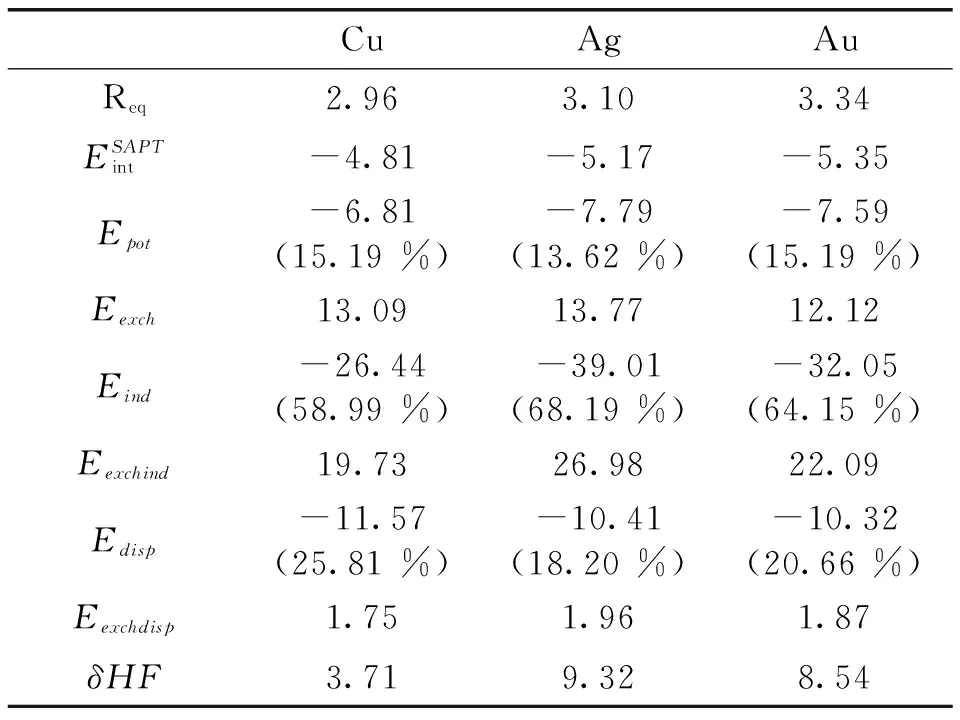
表3 SAPT的相互作用能及其能量分解 / (kcal·mol-1)Tab.3 SAPT interaction energies and their decomposition / (kcal·mol-1)
通过改变二聚体分子间距离RM-M得到SAPT各能量成分见图2。对[Cl-M-PH3]2(M=Cu,Ag,Au)三种体系的静电能(Epot)、诱导能(Eind)和色散能(Edisp)三项均是吸引作用,而交换能(Eexch)、交换-诱导能(Eexch-ind)、交换—色散能(Eexch-disp)和δHF项都是排斥作用。主要是为了估算高级别的诱导和交换—诱导能,在这里不作过多的讨论。
[Cl-Cu-PH3]2中诱导能是主要的吸引作用,在分子平衡距离Req(Cu-Cu)=2.96Å时,诱导相互作用占总吸引能的58.99 %;随着分子间的距离RCu-Cu的增加,诱导相互作用逐渐减小, RCu-Cu等于3.75Å时,色散相互作用和诱导相互作用基本相等,当RCu-Cu大于3.75Å,色散相互作用占主导,例如在RCu-Cu等于4.00Å时,色散能占吸引作用的52.90 %,而诱导能占吸引作用的38.71 %。在RCu-Cu小于2.60Å时,静电相互作用比色散能大,随着分子距离的增加,色散能逐渐大于静电相互作用。[Cl-Cu-PH3]2分子中的排斥作用主要来自于交换-诱导能和交换能。
在[Cl-Ag-PH3]2和[Cl-Au-PH3]2分子中,SAPT各能量成分的变化趋势与[Cl-Cu-PH3]2基本相同。在二聚体分子平衡距离Req附近,诱导能是主要的吸引作用,其次色散能,静电能最小;随着分子间RM-M(M=Cu,Ag,Au)的增加,色散能逐渐占主导作用,这主要是因为在诱导能中起主导作用的多极部分与1 / R8成正比,而色散能与1 / R6成正比,因此诱导能随着分子间距离的增加迅速下降,而色散能下降较平缓;当两分子的金属在较近的距离时,静电能比色散能大,随着分子间距离RM-M的增加,色散能逐渐大于静电能。
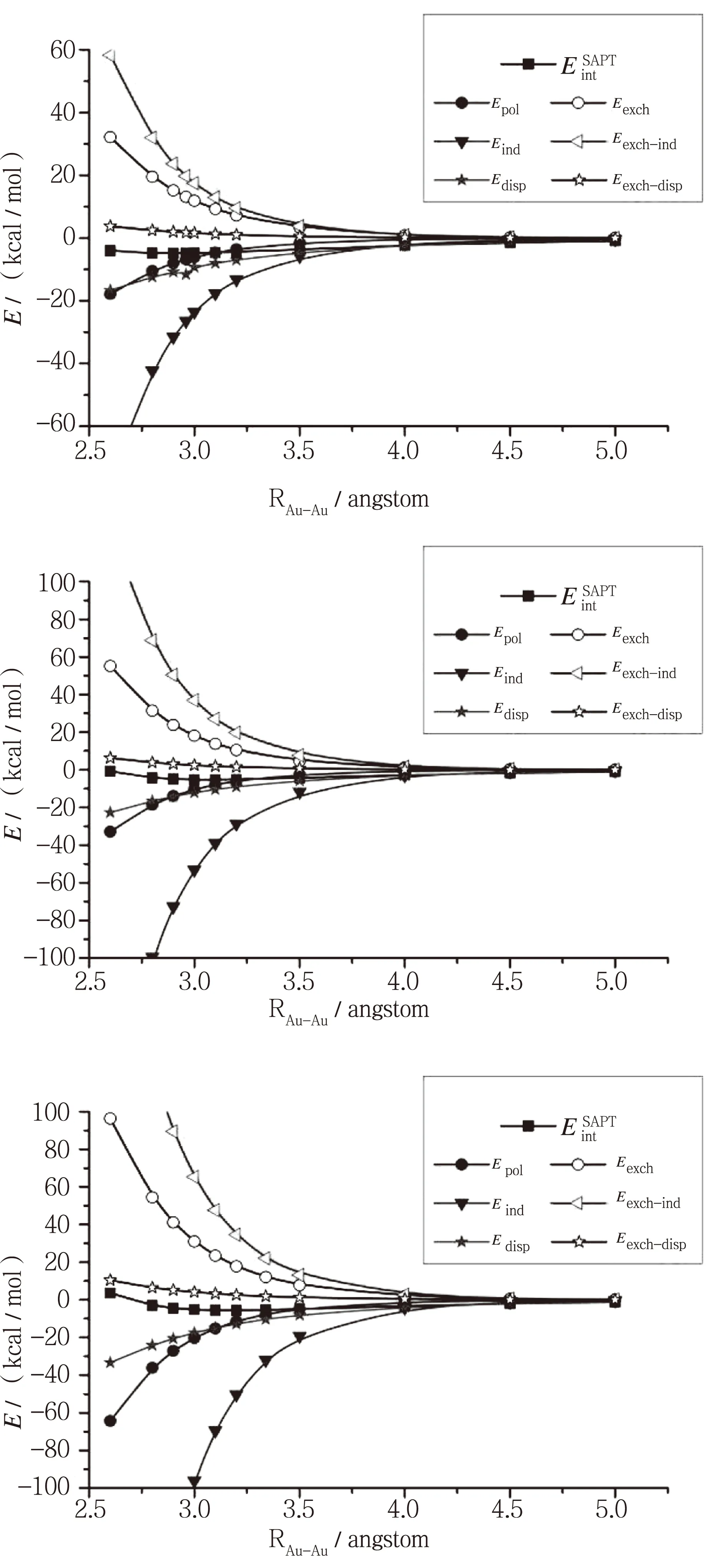
图2 [Cl-M-PH3]2(M=Cu,Ag,Au)的SAPT相互作用 能及其各能量成分与分子间距离RM-MFig.2 The SAPT interaction energy decomposition for the [Cl-M-PH3]2(M=Cu,Ag,Au)at different intermolecular distance RM-M
如图3所示[Cl-M-PH3]2(M=Cu,Ag,Au)三种体系的静电能、诱导能和色散能随分子间相互作用距离RM-M变化的情况,有图3可以看出随着原子序数的增加,由Cu到Au静电能、诱导能和色散能均逐渐增大,由Cu到Au相互作用能逐渐增大(SAPT计算的相互作用能由Cu到Au逐渐增大),但是因为随分子间距离RM-M的增大诱导能迅速下降,静电能、色散能也逐渐减小,但是变化较平缓;[Cl-M-PH3]2(M=Cu,Ag,Au)平衡距离RM-M也逐渐增大(Cu:2.96Å;Ag:3.10Å;Au:3.34Å),所以在平衡距离时的稳定结构,[Cl-Ag-PH3]2的诱导能最大(-39.01 kcal·mol-1),占吸引作用的68.19 %,而[Cl-Au-PH3]2的诱导能(-32.05 kcal·mol-1)只占吸引作用的64.15 %;三种体系在平衡结构时的静电能和色散能差别不大。由以上分析可以看出亲金属相互作用中诱导相互作用是主要吸引作用,而静电相互作用和色散力加强了这种作用。CCSD(T)计算结果为 [Cl-Ag-PH3]2的相互作用能最大,就是因为其在平衡结构时有较大的诱导能。
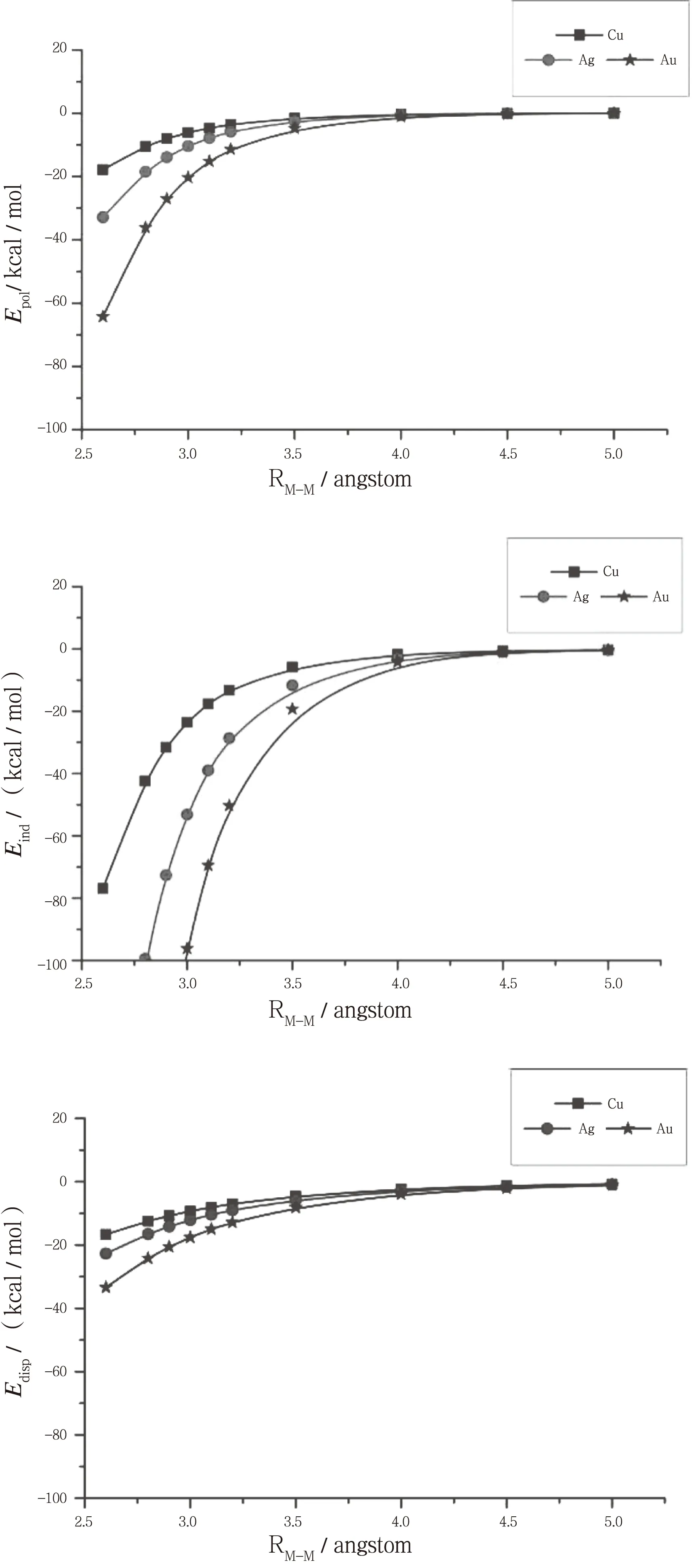
图3 [Cl-M-PH3]2(M=Cu,Ag,Au)中静电能、 诱导能和色散能与分子间距离RM-M的关系Fig.3 Relationship between electrostatic energy、induction energy and dispersion energy with intermolecular distance RM-M of [Cl-M-PH3]2(M=Cu,Ag,Au)
Mangnko等人使用LMP2计算[Cl-M-PH3]2(M=Cu,Ag,Au)相互作用能分别为-5.28 kcal·mol-1,-7.34 kcal·mol-1,-8.32 kcal·mol-1,并进行能量分解,在平衡结构的结果表明色散力占吸引作用的60 %以上,离子激发也是主要的吸引作用[10]。与CCSD(T)结果相比,LMP2过高的计算了分子间相互作用,SAPT计算结果表明[Cl-M-PH3]2(M=Cu,Ag,Au)亲和作用诱导起主导作用,而不是色散作用。
3 结论
使用CCSD(T)对[Cl-M-PH3]2(M=Cu,Ag,Au)的亲金属效应进行理论研究,结果表明[Cl-M-PH3]2(M=Cu,Ag,Au)的亲金属相互作用能整体差别不大,在-5 kcal·mol-1左右,与二聚水氢键键能大小相当。亲银作用较强,亲金次之,亲铜较弱。SAPT对[Cl-M-PH3]2(M=Cu,Ag,Au)计算结果表明由Cu到Au的静电能,诱导能,色散能均逐渐增加。诱导能在亲金属相互作用中占主导作用,静电能和色散能加强了这种相互作用。[Cl-Ag-PH3]2的诱导能最大,三种二聚体的静电能和色散能相差不大。
【REFERENCES】
[1]HEINE A,Herbst-Irmer R,Stalke D.[Cu2R2BrLi(thf)3],R = Si(SiMe3)3-a complex containing five-coordinate silicon in a three-centre two-electron bond(thf = tetrahydrofura-n)[J].J.Chem.Soc.Chem.Commun.,1993,23:1729-1731.
[2]Schmidbaur H.Ludwig mond lecture.High-carat Gold Cu-omponds [J].Chem.Soc.Rev.,1995,24:391-400.
[3]PykkÖ P.Strong Closed-Shell Interactions in Inorganic Chemistry [J].Chem.Rev.,1997,97(3):597-636.
[4]OMARY M A,Webb T R,Assefa Z,et al.Crystal Structure,Electronic Structure,and Temperature-Dependent Raman Spectra of Tl[Ag(CN)2]:Evidence for Ligand-Unsupported Argentophilic Interactions [J].Inorg.Chem.,1998,37(6):1380-1386.
[5]麦松威,周公度,李伟基.高等无机结构化学[M].北京:北京大学,2001:489-496.
MAI S W,ZHOU G D,LI W J.Advanced Structural Inprganic Chemistry[M].Beijing:Peking University,2001:489-496.
[6]PYKKÖ P,ZHAO Y F.Ab initio Calculations on the(ClAuPH3)2Dimer with Relativistic Pseudopotential:Is the “A-urophilic Attraction” a Correlation Effect?[J].Angew.Chem.,Int.Ed.Engl.,1991,30(5):604-605.
[7]PYKKÖ P,LI J,Runeberg N.Predicted ligand dependen-ce of the Au(I)…Au(I)attraction in(XAuPH3)2[J].Chem.Phys.Lett.,1994,218(1-2):133-138.
[8]PYKKÖ P,RUNE B N,Mendizabal F.Theory of the d10-d10Closed-Shell Attraction:1.Dimers Near Equilibrium[J].Chem.-Eur.J.,1997,3(9):1451-1457.
[9]RUNE B N,Schütz M,Werner H-J.The aurophilic attraction as interpreted by local correlation methods[J].J.Chem.Phys.,1999,110(15):7210-7215.
[10]MAGNKO L,SCHWEIZER M,Rauhut G,et al.A comparison of metallophilic attraction in(X-M-PH3)2(M=Cu,Ag,Au;X=H,Cl)[J].Phys.Chem.Chem.Phys.,2002,4(6):1006-1013.
[11]O’GRADY E,Kaltsoyannis N.Does metallophilicity increase or decrease down group 11? Computational investigations of [Cl-M-PH3]2(M=Cu,Ag,Au,[111])[J].Phys.Chem.Chem.Phys.,2004,6(4):680-687.
[12]DUN N Jr T H.Gaussian basis sets for use in correlated molecular calculations.I.The atoms boron through neon and hydrogen[J].J.Chem.Phys.,1989,90(2):1007-1023.
[13]WOON D E,DUNNNING Jr T H.Gaussian basis sets for use in correlated molecular calculations.III.The atoms aluminum through argon[J].J.Chem.Phys.,1993,98(2):1358-1371.
[14]PETERSON K A,Puzzarin C.Systematically convergent basis sets for transition metals.II.Pseudopotential-based correlation consistent basis sets for the group 11(Cu,Ag,Au)and 12(Zn,Cd,Hg)elements [J].Theor.Chem.Acc.,2005,114(4):283-296.
[15]FIGGEN D,RAUHUT G,DOLG M,et al.Energy-consistent pseudopotentials for group 11 and 12 atoms:adjustment to multi-configuration Dirac-Hartree-Fock data[J].Chem.Phys.,2005,311(1-2):227-244.
[16]BOYS S F,BERNARDI F.The calculation of small molecular interactions by the differences of separate total energies.Some procedures with reduced errors[J].Mol.Phys.,1970,19(4):553-566.
[17]JEZIORSKI B,MOSZYNSKI R,Szalewicz K.Perturbati-on Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes[J].Chem.Rev.,1994,94(7):1887-1930.
[18]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09[CP].Wallingford:Gaussian,Inc.,2009.
[19]WERNER H J,KNOWLES P J,LINDH R,et al.MOL-PRO[CP].2012.
[20]BOWMAKER G A,SCHMIDBAUR H, Krüger S,et al.A Density Functional Study of Metal?Ligand Bonding in [(PR3)2M]+ and [PR3MCl](M = Ag,Au;R = H,Me)Complexes[J].Inorg.Chem.,1997,36(9):1754-1757.
[21]BAENZIGER N C,BENNETT W E,SOBOROFF D M.Chloro(triphenylphosphine)gold(I)[J].Acta Cryst.Sect.B.,1976,32:962-963.[22]ANGERMAIER K,ZELLER E,SCHMIDBAUR H.Cryst-al structures of chloro(trimethylphosphine)gold(I),chloro(tri-ipropylphosphine)gold(I)and bis(trimethylphosphine)gold(I)chloride[J].J.Organomet.Chem.,1994,472(1-2):371-376.
[23]TSUZUKI S,HONDA K,UCHIMARU T,et al.The Magnitude of the CH / π Interaction between Benzene and Some Model Hydrocarbons[J].J.Am.Chem.Soc.,2000,122(15):3746-3753.
[24]SINNOKROT M O,SHERRILLl C D.Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer:Sandwich,T-Shaped,and Parallel-Displaced Configurations[J].J.Phys.Chem.A.,2004,108(46):10200-10207.
[25]PATKOWSKI K,PODESZWA R,SZALEWICZ K.Inter-actions in Diatomic Dimers Involving Closed-Shell Metals[J].J.Phys.Chem.A.,2007,111(49):12822-12838.
Theoretical study on the metallophilic interactions of the [Cl-M-PH3]2(M=Cu,Ag,Au)complexes
SUN Tao1,2,WANG Yibo1,2▲
(GuizhouKeyLaboratoryofHighPerformanceComputationalChemistry,NetworkandInformationCenter,GuizhouUniversity,Guiyang550025,China)
The metallophilic interactions of the [Cl-M-PH3]2complexes(M = Cu,Ag,Au)were studied using the CCSD(T)/ aug-cc-pVTZ high level electronic correlation theory.The results showed that there were small differences among the interaction energies,with argentophilic attraction being the strongest,the aurophilic attraction taking second place and the cuprophilic attraction being the weakest.The symmetry adapted perturbation theory SAPT was adopted to analyze the energy decomposition,and the results showed that the electrostatic interaction,induction interaction and dispersion interaction increased gradually from Cu to Au.The induction interaction,which accounted for over 58 % of the main attraction interaction,played a leading role.The electrostatic interaction and dispersion interaction did not show much difference.The induction interaction of [Cl-Ag- PH3]2was stronger than that of [Cl-Au- PH3]2.
metallophilic interaction,intermolecular interaction,SAPT,energy decomposition
O641
A
1003-6563(2016)04-0064-06
2016 -06-02;
无
孙涛(1982-),男,实验师,主要从事分子间相互作用理论研究。
▲王一波(1962-),男,教授,主要从事量子化学和分子间相互作用理论研究。
