烯胺合成的研究进展
2016-08-10王艳红黄文华
王艳红,黄文华
(天津大学化学系,天津 300072)
烯胺合成的研究进展
王艳红,黄文华
(天津大学化学系,天津 300072)
摘要:综述了烯胺合成方法的最新进展:酮或醛与胺的缩合法、过渡金属催化的偶联反应法、酮肟或酰胺的还原法、炔烃或腈的加成反应法,介绍了各合成方法的优缺点,对烯胺合成方法的选择具有重要的指导意义。
关键词:缩合;过渡金属催化偶联;还原;加成;烯胺
烯胺是有机合成和药物化学中非常重要的中间体,烯胺对亲电子体具有很好的反应活性,能参与一系列重要官能团间的转变,通常用来进行具有立体选择性的共轭加成、形成杂环化合物和环加成反应[1-2];烯胺也可直接通过还原氨化生成相应的胺或通过不对称氢化生成手性胺[3];烯胺的另一个重要应用是实现羰基化合物的烷基化和酰基化[4-5]。但是,由于反应条件、底物或纯化方法等因素的影响,除了β-羰基共轭烯胺外,其它烯胺通常收率低、稳定性差、操作过程中易分解。因此,很多研究者都在探索新的收率较高、立体选择性好且稳定的烯胺合成方法。
作者对烯胺的最新合成方法进行了综述,拟为进一步加快烯胺化学的发展提供帮助。
1酮或醛与胺的缩合法
Langer等[6]发现了一个快速、温和、立体选择性好的由一系列醛(化合物1)与二级胺(化合物2)发生缩合反应生成二取代或三取代的烯胺化合物3的方法(图1)。探索了一系列二级胺(二苄胺、二乙胺、吗啉、吡咯烷等)与一系列醛化合物的缩合,发现,在0 ℃、4A分子筛催化下,1.2eq.的胺与醛缩合可得到收率为84%~100%的烯胺化合物3,底物醛对于一般的官能团、甚至含有甲硅烷基醚和二甲基缩醛官能团的底物均具有很好的兼容性。该方法既避免了传统方法需使用酸、碱和高温的苛刻条件,同时又能得到高收率的产物。但烯胺本身不稳定,大部分产物只能原位产生使用,为此很多研究者致力于研究以β-羰基共轭形式稳定的烯胺化合物。

图1 醛与二级胺的反应1 The reaction of aldehydes and secondary amines
Sun等[7]在温和条件下用α-溴代芳香酮(化合物4)与N-保护的胺(化合物5)一锅法两步合成了功能化的烯胺化合物7(图2)。首先,N-保护的胺与α-溴代芳香酮发生取代反应生成中间产物6,然后在碱的作用下中间产物6中的苄醇发生消除异构化反应形成α,β-双键,最终得到功能化的单一Z-型烯胺化合物7。溶剂选用对两步均有促进作用的极性非质子型溶剂乙腈;而碱,第一步选用碱性相对较弱的碳酸铯,避免强碱条件下化合物4发生自消除反应,第二步,为缩短反应时间和提高收率,需在强碱DBU作用下,加热回流一段时间。通过该法能得到收率为74%~92%的稳定的β-羰基烯胺化合物。
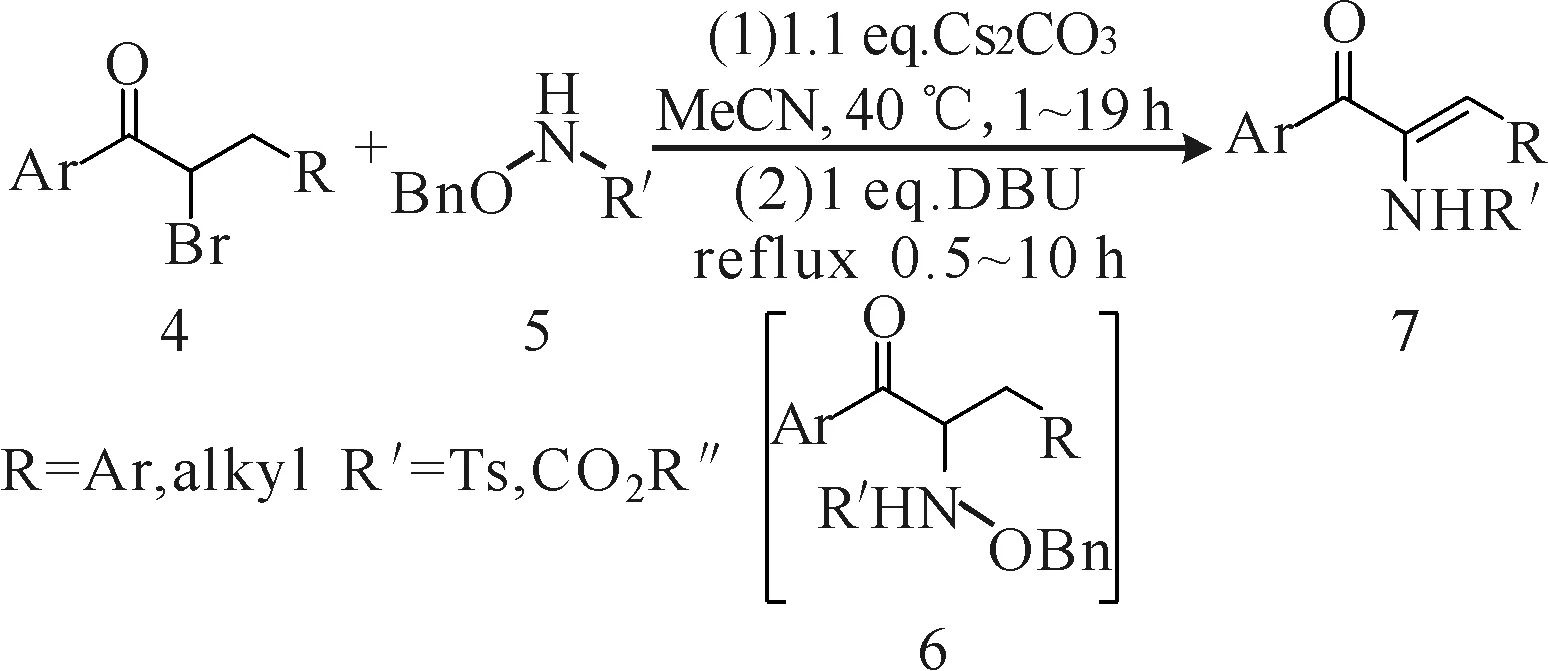
图2 α-溴代芳香酮与N-保护的胺的反应Fig.2 The reaction of α-bromoketones compound and N-protected amines
Xu等[8]改进了传统的酮与二级胺的缩合反应条件,用固体的1, 3-二羰基化合物(化合物8)与芳苯胺(化合物9)在室温、固态条件下发生缩合反应制备烯胺酮类化合物10(图3)。该反应运用机械化学的方法通过催化剂量的硫酸氢钾和二氧化硅的机械化学磨合即可发生,不需加入溶剂、酸或碱,环保安全、操作简单、条件温和且反应时间较短、产物纯净、原子利用率和收率高(90%~99%),且固体1,3-二羰基化合物和固体苯胺类化合物的底物范围较广。
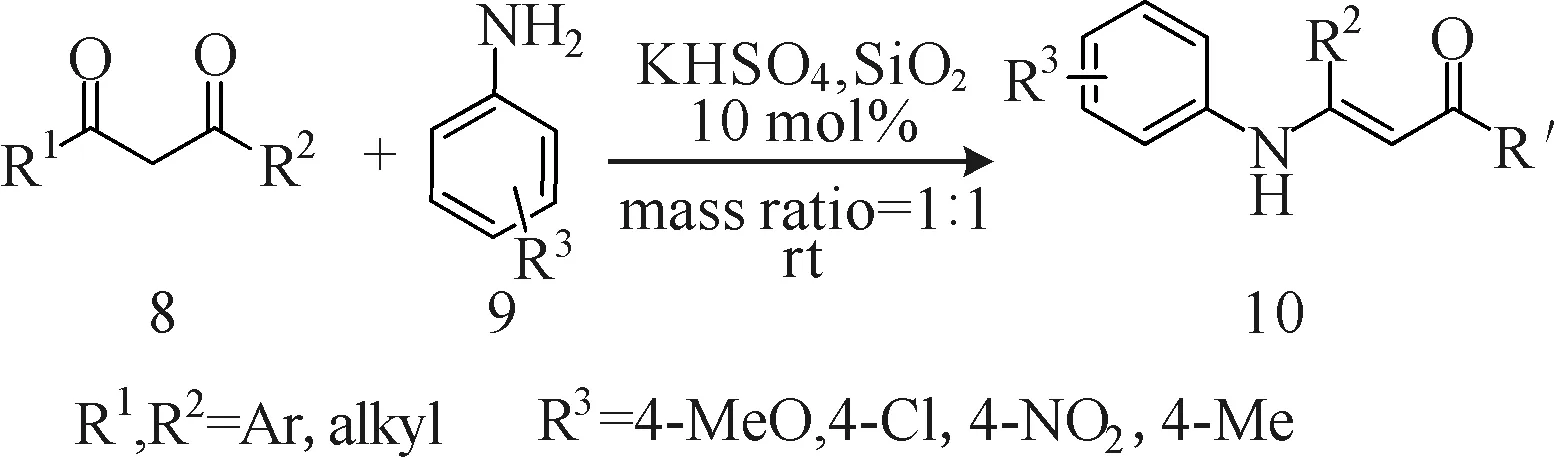
图3 在固态条件下,1,3-二羰基化合物与胺合成 β-烯胺酮化合物Fig.3 Synthesis of β-enamino ketones from 1,3-dicarbonyl compounds and amines under the condition of solid state
2过渡金属催化的偶联反应法
Wang等[9]报道了一种由卤代烯烃(化合物11)和二级胺(化合物12)通过铜催化的偶联反应制备烯胺化合物13的方法(图4)。即以溴代或碘代烯烃及二级胺为原料、稳定廉价的CuI为催化剂、N,N′-二甲基乙烷-1, 2′-二胺为配体、叔丁醇钠为碱,在甲苯溶剂中于140 ℃下加热回流制备烯胺化合物。该方法不仅适用于各种溴代或碘代烷基烯烃与二级芳香胺的偶联反应,也适用于卤代芳香烯烃与二级芳香胺之间的偶联反应,这在铜催化不活泼的溴代烯烃氨基化反应中堪称首例。此法虽然能得到具有较高收率(71%~97%)和较好E/Z比的烯胺化合物,但其对含活泼β-H且易发生E2消除反应的卤代烯烃并不适用,并且反应需高温回流且反应时间较长(16~72 h)。
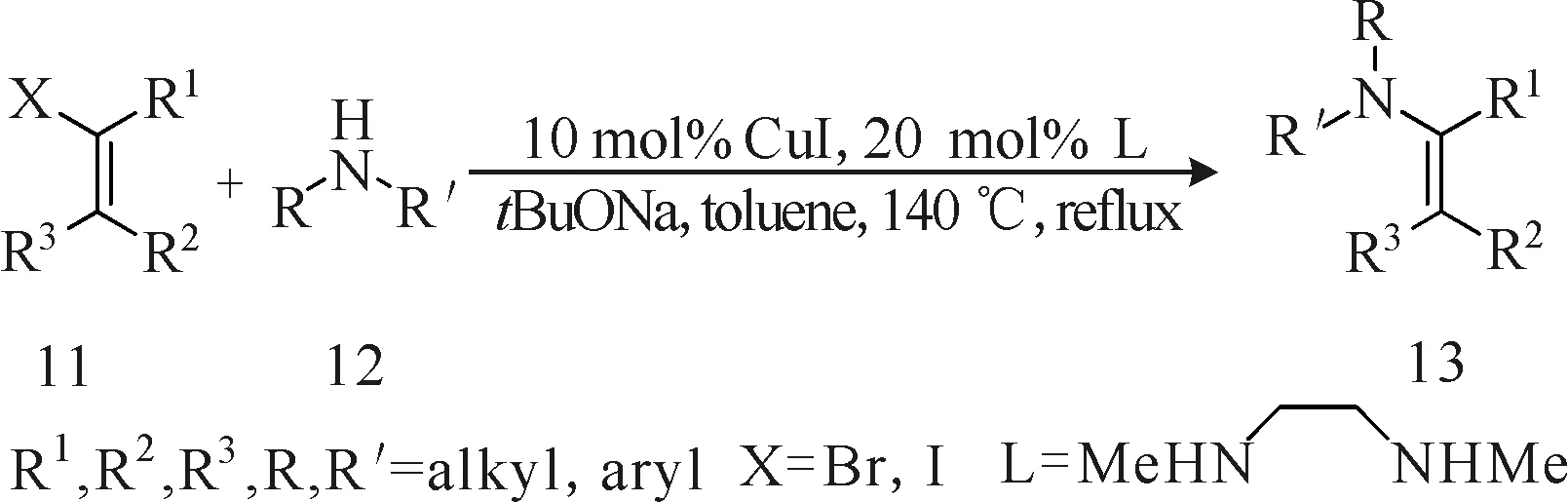
图4 铜催化的卤代烯烃的胺化反应Fig.4 Copper-catalyzed amination of alkenyl halides
Yan等[10]在铜催化卤代烯烃的胺化反应的基础上,对底物进行修饰,克服了需高温和反应时间长的缺点,即设计了在有机合成中常用且易得的烯烃茂锆试剂(化合物14)与邻苯甲酰羟胺(化合物15)的亲电胺化反应(图5)。这是一个合成多取代、双键构型保持和具有空间位阻的烯胺化合物16的较好方法,该方法具有较好的官能团兼容性、反应条件温和、反应快且收率较高,通过简单的萃取和气相分离手段即可得到纯净的产物。但是,当用单取代或二取代的烯烃茂锆试剂与邻苯甲酰羟胺反应时却得到了1,3-二烯化合物,并未得到目标产物,因此该方法不适用于单取代或二取代的烯胺化合物的合成。

图5 铜催化的烯烃茂锆试剂与邻苯甲酰羟胺的亲电胺化反应Fig.5 Copper-catalyzed electrophilic amination of alkenylzirconocenes with o-benzoylhydroxylamine
Shin等[11]将炔烃(化合物17)、N-磺酰叠氮化物(化合物18)和芳香醚衍生物(化合物20)通过铜催化的串联环加成和铑催化的烷氧基化一锅法合成了2-芳烷基-2-芳基-烯胺化合物21(图6)。该化合物具有生物学活性,部分存在于哺乳动物的甲状腺激素中。过渡金属铑催化N-磺酸基-4-芳基-1,2,3-苯三唑(化合物19)插入到芳香醚衍生物(化合物20)的芳香C-H键中,再消除1分子氮气得到目标产物21。化合物19无需分离,可直接与化合物20一锅法生成产物21,且收率较高(70%~90%)。该方法简化了反应步骤。化合物20既是原料又是溶剂,避免了传统方法中有毒溶剂的使用,但同时使用2种过渡金属,成本较高。
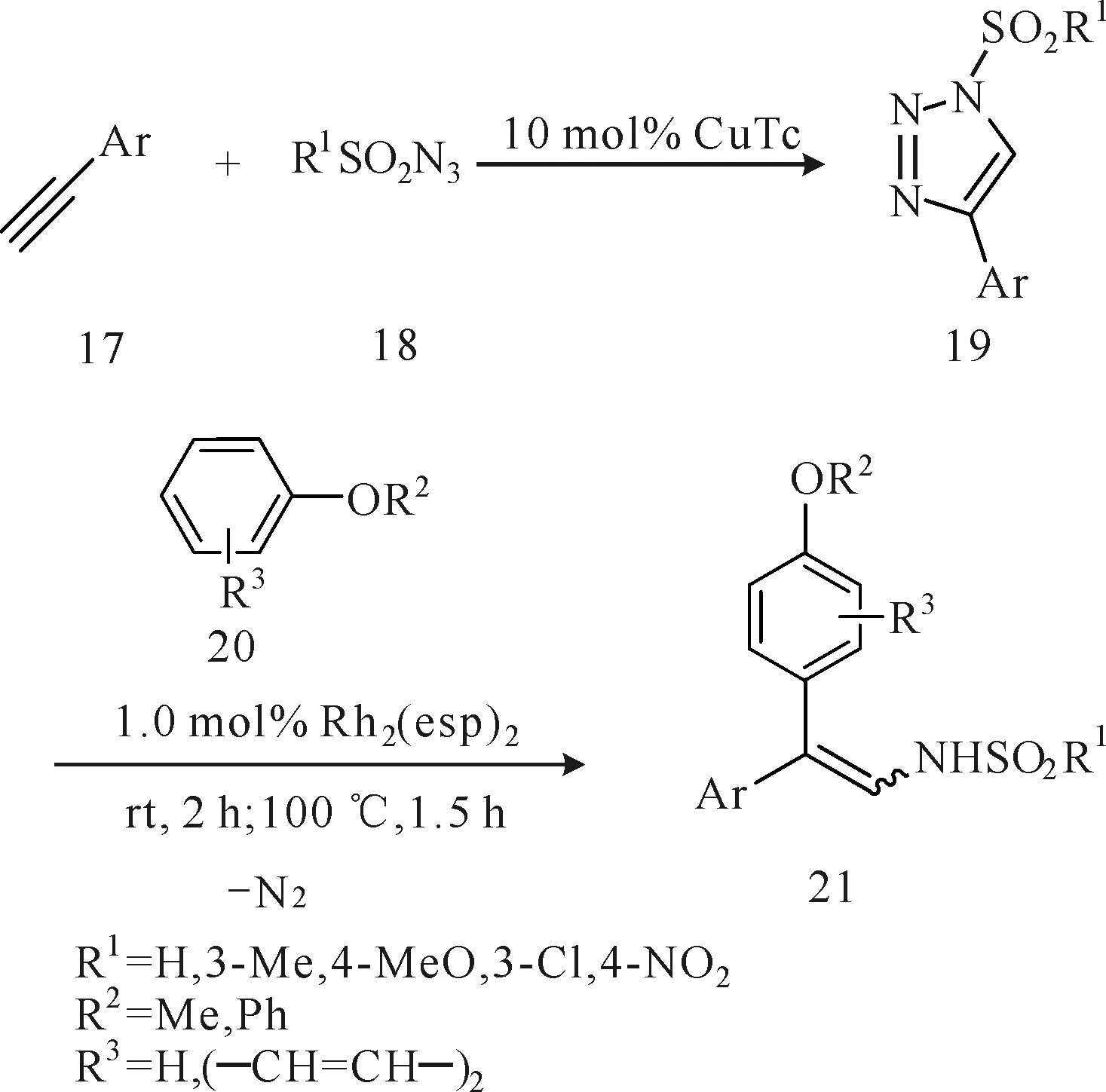
图6 铜催化的炔烃、N-磺酰叠氮化物和芳香醚的串联环加成 反应和铑催化的烷氧基化法合成2-芳烷基-2-芳基-烯胺Fig.6 Synthesis of 2-alkoxyaryl-2-aryl enamines via tandem copper-catalyzed cycloaddition and rhodium-catalyzed alkoxyarylation from alkynes,N-sulfonylazides and aryl ethers
Ji等[12]提出一种单一钯催化剂且钯可以循环使用的催化方法。即在金属钯和1分子氧气存在的条件下,取代的芳香一级胺类衍生物(化合物22)与烯烃衍生物(化合物23)发生氧化偶联反应快速合成立体选择性的单一Z-型(Z/E>99∶1)烯胺化合物24(图7)。首先Pd(Ⅱ)与化合物23配位,然后亲核试剂化合物22进攻此配合物产生具有分子内氢键的σ-烷基钯复合物,此复合物随即发生β-H的消除反应,释放出化合物24和Pd(0),Pd(0)经氧气氧化生成Pd(Ⅱ),以此开始催化循环。而LiBr在该反应中起着阻止化合物22与钯配位的作用。该法中底物22和23均具有极好的官能团兼容性且便宜易得,而水作为此反应的唯一副产物,提高了原子利用率,收率最高可达97%,且分离操作简单,此外,产物烯胺具有很高的反应活性,易转化为一系列含氮杂环化合物,在有机合成和医药化工领域具有巨大的应用价值。
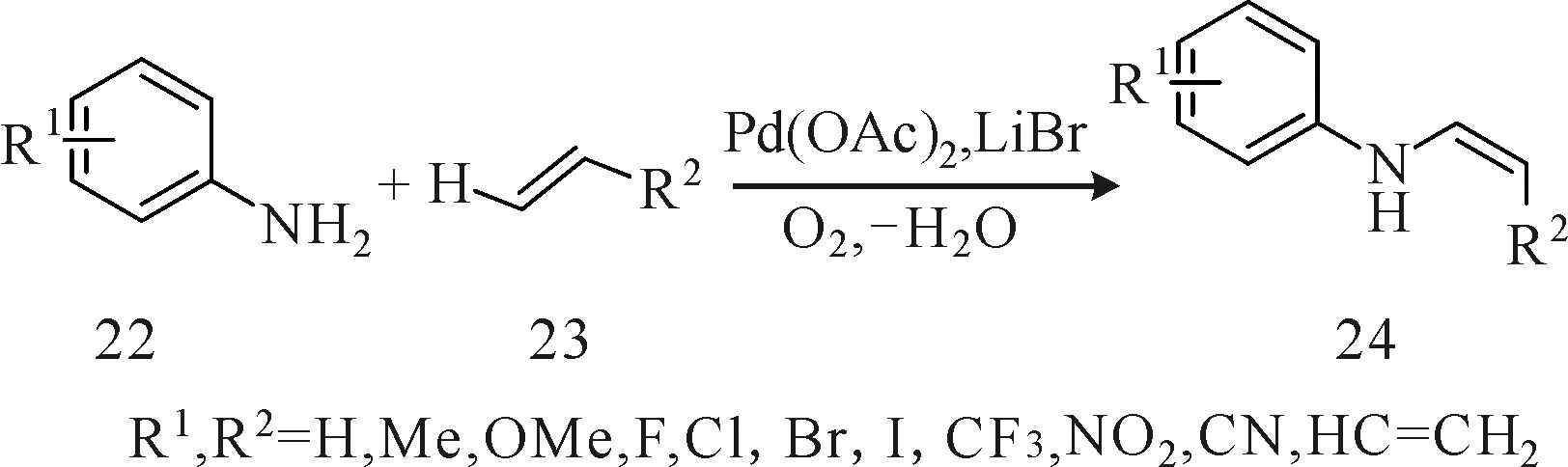
图7 钯催化芳香一级胺化合物与烯烃化合物氧化偶联Fig.7 Palladium-catalyzed oxidative coupling of aromatic primary amine and alkenes
3酮肟或酰胺的还原法
Tang等[13]报道了酮肟衍生物(化合物25)一步合成N-乙酰-烯胺化合物26的方法(图8)。以Fe(OAc)2为催化剂,使N-O键在温和条件下发生还原裂解形成活泼的亚胺,再经乙酸酐猝灭酰化,最后经纯化得收率较高(66%~90%)的烯胺。与其它金属催化相比,乙酸亚铁催化是最常用、最经济的且对各种官能团均具有兼容性的催化方法,是一个比较方便实用的N-乙酰-烯胺合成方法,也为工业上经不对称催化N-乙酰-烯胺合成手性胺提供了参考,但即使少量的乙酸亚铁残存也会影响烯胺产物的后续应用,且E/Z选择性不佳(E/Z≈1∶1.5),无法得到单一构型的烯胺。

图8 N-乙酰-烯胺的合成Fig.8 Synthesis of N-acetyl enamines
Zhao等[14]报道了一种以酮为起始原料经两步制备烯胺的方法(图9)。酮(化合物27)先与羟胺盐酸盐形成酮肟(化合物28),再在亲氧试剂烷基膦、猝灭剂乙酸酐存在下,于甲苯溶剂中加热回流,最终得到烯胺化合物29。在甲苯加热回流条件下,酮肟的N-O键断裂,而在体系中加入具有亲氧性的烷基膦能促使这一过程加速进行,且产生易清除的水溶性烷基氧膦,底物范围较宽,苄基、烷基、芳香基及非环状的酮均有很好的兼容性且产物烯胺29收率在89%以上。该法以有机亲氧试剂作为还原剂,避免了使用Fe(Ⅱ)盐带来的弊端,为烯胺合成提供了一条很好的途径。
针对产物E/Z选择性不佳的弊端,Volkov等[15]报道了苄基或脂肪族的三级酰胺衍生物(化合物30)

图9 酮经两步制备烯胺的合成反应Fig.9 Synthesis of enamines from ketones via two steps
在氢化硅烷化条件下催化还原脱水制备烯胺化合物31(图10)。选用叔丁醇钾为催化剂,以三甲氧基硅烷或三乙氧基硅烷为氢化源,同时避免使用过渡金属催化剂。该方法首先用叔丁醇钾活化硅烷形成五配位的配合物,而此配合物能促使氢化硅转移至酰胺的羰基上形成四配位化合物,随后再发生硅醚的消除和α-质子的迁移,最终形成烯胺化合物31。该方法具有较好的催化效果,可得到单一E-型烯胺化合物,收率为60%~91%,但该反应过程较复杂,反应时间较长,不适合工业化生产。
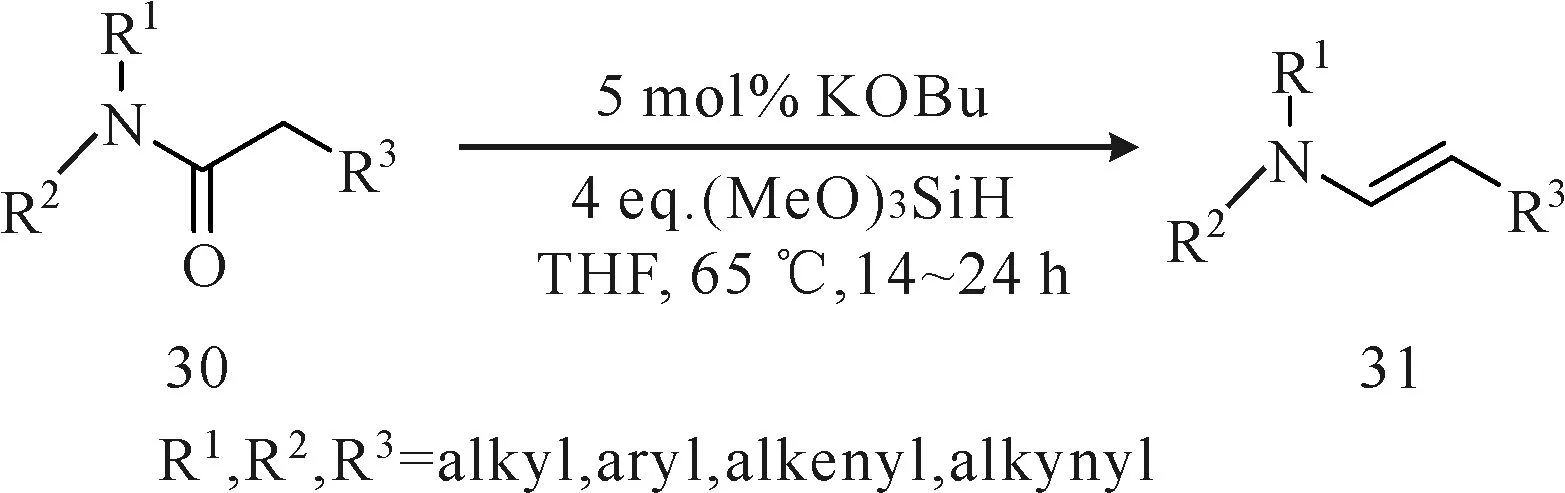
图10 三级酰胺在氢化硅烷化条件下催化还原脱水合成烯胺Fig.10 Synthesis of enamines by catalytic reductive dehydration of tertiary amides under hydrosilylation condition
为了简化酰胺还原为烯胺的过程,Tahara等[16]在过渡金属中设计合适的配体来催化酰胺衍生物(化合物32)还原为烯胺化合物33,即设计合适的配体L,使IrCl(CO)L2能够高效催化合成具有电子转移特性的π-共轭的烯胺(图11)。铱配合物含有吸电子的磷配体,能和氢化硅烷共同催化酰胺的氢化硅烷化,产生相应的硅烷半缩醛胺,再在加热或酸性条件下转化为烯胺。该配合物具有较高的催化活性(TON>10 000)且在烯胺产物中铱残余率低(<20×10-9)。该方法克服了通过醛或酮与二级胺缩合合成π-共轭的烯胺方法中可逆、高温、反应过程中除水难和收率偏低等缺点[17-18]。而与乙烯基卤化物与二级胺的交叉偶联[19-21]、胺脱氢[22]、炔烃的氢胺化反应[23-24]等合成π-共轭的烯胺的方法相比,该法将酰胺直接还原为烯胺,且酰胺易得,催化剂的催化活性较高、反应时间较短(室温条件下30 min即可催化完全)且能得到较好选择性(E/Z>90∶10)的产物。
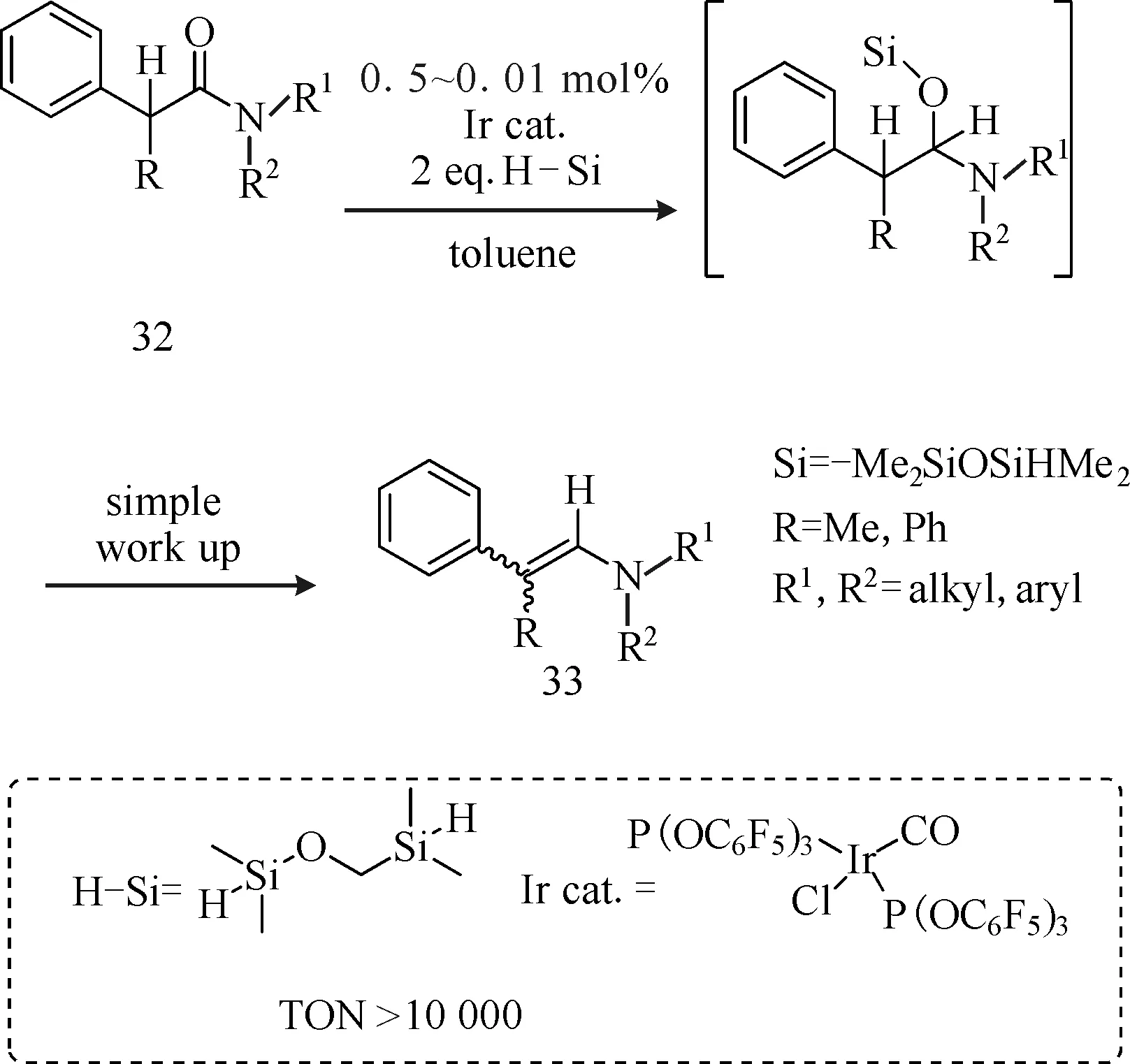
图11 IrCl(CO)L2催化酰胺直接还原为π-共轭的烯胺Fig.11 Synthesis of π-conjugated enamines from amides catalyzed by IrCl(CO)L2
4炔烃或腈的加成反应法
炔烃或腈类化合物的亲电或亲核加成反应法也是合成烯胺化合物的一种常用方法。Savarin等[25]在传统的由腈类化合物与格氏试剂合成烯胺的基础上进行了创新,采用一种可循环使用的混合试剂[有机金属化合物甲基锂和无机金属化合物溴化锂(反应过程中抑制亚胺异构化)]与芳香腈类化合物34在低温条件下反应合成芳香烯胺化合物35(图12)。首先芳香腈底物(吸电子或推电子)与甲基锂发生加成反应生成亚胺,亚胺再经乙酸酐猝灭得到粗产物烯胺,最后经乙醇-水重结晶后得到纯净且收率较高(60%~85%)的N-乙酰烯胺,该方法中混合试剂的使用避免了以往因格氏试剂的加入而使反应体系变复杂的缺点。

图12 有机金属化合物与金属溴化物的混合试剂与腈类反应合成烯胺Fig.12 Synthesis of enamines by reaction of organometallics and metal bromides with nitriles
原位生成布莱斯反应中间体也是合成烯胺化合物的一种方法。Chun等[26]报道,腈类(化合物36)与混合试剂(锌粉和溴代乙酸乙酯)反应原位生成布莱斯反应中间体,然后与各类非活化的末端炔烃或中间炔烃(化合物37)在温和条件下发生亲核加成反应生成α-乙烯化的β-烯胺酯化合物38(图13),收率最高可达91%。在此反应中布莱斯反应中间体起着路易斯酸和有机亲核试剂的双重作用,避免了路易斯酸的使用。同年,Chun等[27]利用此方法合成了烯胺酮类化合物39(图14),收率最高可达98%。首先由Reformatsky试剂和腈类化合物反应原位生成布莱斯反应中间体,然后再与丙炔酸酯衍生物以具有化学和立体选择性的方式发生反应合成2-吡啶酮衍生物,此反应以布莱斯反应中间体作为碳亲核试剂进攻炔烃形成α-乙烯化的β-烯胺酯类化合物[28-29]。该法具有底物(芳香的、杂环的和脂肪族的腈类化合物)范围较宽、收率高的优点,为直接采用腈类化合物来构建各类吡啶酮衍生物提供了一条极好的路线。
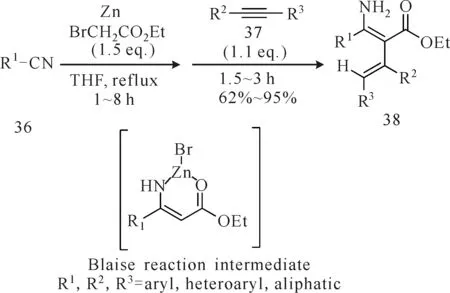
图13 腈类、锌粉和溴代乙酸乙酯的混合试剂与炔烃反应合成α-乙烯化的β-烯胺酯Fig.13 Synthesis of α-vinylated β-enaminoesters from alkyne by the reaction of alkyne,zinc and ethyl bromoacetate
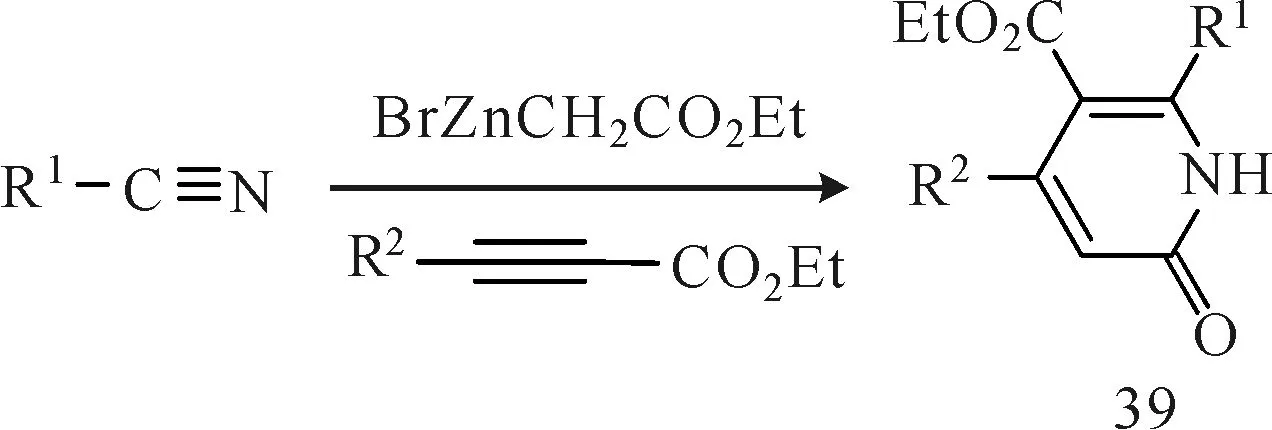
图14 腈类化合物通过一锅法合成烯胺酮类化合物Fig.14 One-pot synthesis of enaminone from nitriles
Lee等[30]报道了一种新的现场生成有机金属试剂的温和方法,即在氯化锌和Hünig′s碱(N,N-二异丙基乙胺)的存在下,腈类化合物(化合物40)与丙二酸单乙酯钾盐发生反应,可得到收率较高的β-氨基丙烯酸酯化合物41(图15)。首先氯化锌与丙二酸单乙酯钾盐形成配合物,然后此配合物在Hünig′s碱的作用下发生脱羧反应,现场生成有机锌试剂,再与腈类化合物发生加成反应生成化合物41。该反应为吸热反应,比经典的布莱斯反应更加安全,且避免了催泪试剂溴乙酸盐的使用,催化剂氯化锌试剂的用量也较少(0.5~1 eq.),便于后续残留物的消除。但底物腈类化合物的反应活性与取代基的电子效应和空间效应有关,如,取代基为吸电子的腈反应活性高于取代基为给电子的腈;对位取代的腈反应活性高于邻位取代的腈;杂环和环丙基腈反应效果较好,但都存在反应时间较长(18~72 h)的缺点。
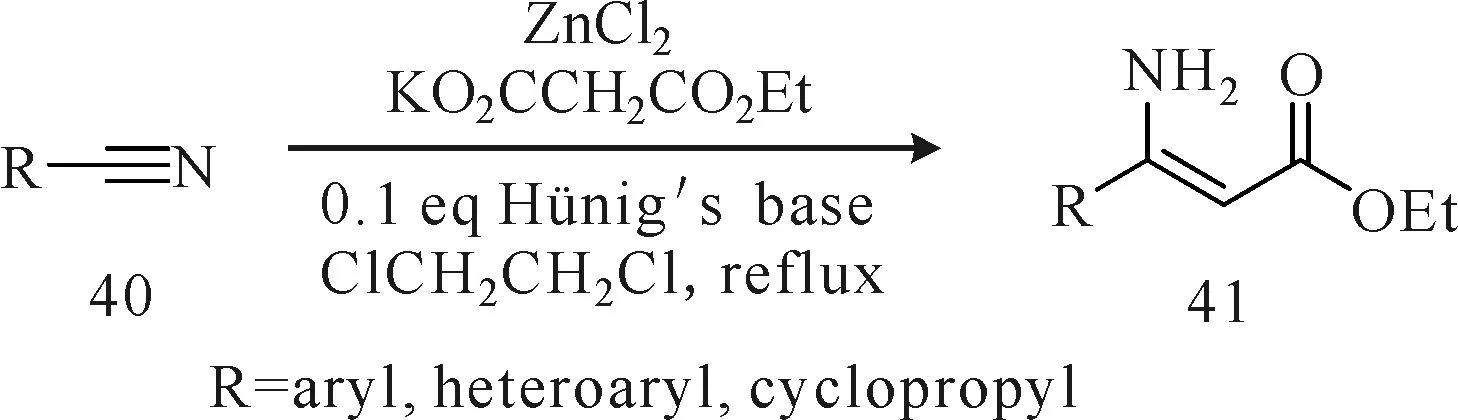
图15 氯化锌和Hünig′s碱催化腈类与丙二酸单乙酯钾盐反应合成β-氨基丙烯酸酯Fig.15 Synthesis of β-amino acrylates by reaction of nitriles with potassium ethyl malonate in the presence of zinc chloride and Hünig′s base
在球磨机中进行反应是合成化学研究的一个新兴领域,其适用于很多有机化学反应类型,如羟醛型反应、氧化反应、还原反应、金属催化的新键形成反应,且在缩短反应时间和提高收率上具有显著效果。Thorwirth等[31]报道了在无溶剂体系条件下的球磨机中,由烷基丙炔酸酯化合物与胺快速合成烯胺化合物的方法(图16)。在无任何催化剂、酸或碱的条件下,该反应在球磨机中5 min即可将烷基丙炔酸酯(化合物42)和胺(化合物43)等当量地转化成烯胺化合物44,且无任何副产物产生,但是得到的是E、Z构型混合底物。E/Z比与底物类型有关,比如二级烷基胺形成单一E-型烯胺产物,而苯胺化合物形成E/Z为97∶3的异构体混合物。该法具有收率较高(96%)和反应时间较短的优点。

图16 烷基丙炔酸酯合成烯胺化合物Fig.16 Synthesis of enamines from alkyl ester of propiolic acid
5结论
4种合成烯胺的方法都具有反应条件温和、选择性好、催化效率高和收率较高的优点。除缩合法外,过渡金属催化法、还原法和加成法合成烯胺的底物范围广;而过渡金属催化法因过渡金属成本较高,反应体系中有金属残余等不适用于放大反应,因此还原法和加成法是今后烯胺合成的研究方向。可以预见,随着研究的不断深入,烯胺化学会取得更大的发展,烯胺的应用也会更加广泛。
参考文献:
[1]HANADA S,TSUTUMI E,MOTOYAMA Y,et al.Practical access to amines by platinum-catalyzed reduction of carbonamides with hydrosilanes:synergy of dual Si-H groups leads to high efficiency and selectivity[J].J Am Chem Soc,2009,131(41):15032-15040.
[2]ZHU W M,LI Z F,ZHANG Y M.Synthesis of enaminesviathe cross-coupling of thioamides and diarylketones promoted by the Sm/SmI2mixed reagent[J].Chinese Chem Lett,2006,17(1):1-4.
[3]HOU G H,XIE J H,YAN P C,et al.Iridium-catalyzed asymmetric hydrogenation of cyclic enamines[J].J Am Chem Soc,2008,131:1366-1367.
[4]STORK G,TERRELL R,SZMUSZKOVICZ J.Synthesis of 2-alkyl and 2-acyl ketones[J].J Am Chem Soc,1954,76:2029-2030.
[5]STORK G,LANDESMAN H K.A new ring-enlargement sequence[J].J Am Chem Soc,1956,78:5129-5130.
[6]LANGER G B,DORE M,MENARD F,et al.Highly chemoselective formation of aldehyde enamines under very mild reaction conditions[J].J Org Chem,2006,71:7481-7484.
[7]SUN Y,WANG X Z,ZHANG X F,et al.Synthesis of functionalized enamines:a facile and efficient protocol towardN-protectedα,β-dehydroamino acid derivatives[J].Synlett,2008(6):861-866.
[8]XU S L,LI C P,LI J H.Solid-state synthesis ofβ-enamine ketones from solid 1,3-dicarbonyl compounds and ammonium salts or amines[J].Synlett,2009(5):818-822.
[9]WANG Y X,LIAO Q,XI C Y.Copper-catalyzed amination of alkenyl halides:efficient method for the synthesis of enamines[J].Org Lett,2010,12(13):2951-2953.
[10]YAN X Y,CHEN C,ZHOU Y Q,et al.Copper-catalyzed electrophilic amination of alkenylhydroxylamines witho-benzoylhydroxylamines:an efficient method for synthesis of enamines[J].Org Lett,2012,14(18):4750-4753.
[11]SHIN S,PARK Y,KIM C E,et al.Synthesis of 2-alkoxyaryl-2-aryl enaminesviatandem copper catalyzed cycloaddition and rhodium catalyzed alkoxyarylation from alkynes,N-sulfonyl azides and aryl ethers[J].J Org Chem,2015,80:5859-5869.
[12]JI X C,HUANG H W,WU W Q,et al.Palladium-catalyzed oxidative coupling of aromatic primary amines and alkenes under molecular oxygen:stereoselective assembly of (Z)-enamines[J].J Org Chem,2013,78:11155-11162.
[13]TANG W J,CAPACCI A,SARVESTANI M,et al.A facile and practical synthesis ofN-acetyl enamines[J].J Org Chem,2009,74:9528-9530.
[14]ZHAO H,CHARLES P,VANDENBOSSCHE S G,et al.An efficient synthesis of enamines from ketones[J].Org Lett,2008,10(3):505-507.
[15]VOLKOV A,TINNIS F,ADOLFSSON H.Catalytic reductive dehydration of tertiary amines to enamines under hydrosilylation conditions[J].Org Lett,2014,16:680-683.
[16]TAHARA A,MIYAMOTO Y,AOTO R,et al.Catalyst design of vaska-type iridium complexes for highly efficient synthesis ofπ-conjugated enamines[J].Organometallics,2015,34:4895-4907.
[17]MÜLLER T E,BELLER M.Metal-initiated amination of alkenes and alkynes[J].Chem Rev,1998,98(2):675-704.
[18]KATRITZKY A R,LONG Q H,LUE P,et al.Benzotrizole-assisted synthesis of enamines[J].Tetrahedron,1990,46(24):8153-8160.
[19]VENKAT R C R,URGAONKAR S,VERKADE J G.A highly effective catalyst system for the Pd-catalyzed amination of vinyl bromides and chlorides[J].Org Lett,2005,7(20):4427-4430.
[20]DEHLI J R,LEGROS J,BOLM C.Synthesis of enamines,enol ethers and related compound by cross-coupling reactions[J].Chem Commun,2005(8):973-986.
[21]BARLUENGA J,FERNANDEZ M A,AZNAR F,et al.Palladium catalyzed cross-coupling reactions of amines with alkenyl bromides:a new method for synthesis of enamines and imines[J].Chem Eur J,2004,10(2):494-507.
[22]ZHANG X,FRIED A,KNAPP S,et al.Novel synthesis of enamines by iridium-catalyzed dehydrogenation of tertiary amines[J].Chem Commun,2003(16):2060-2061.
[23]FUKUMOTO Y,ASAI H,SHIMIZU M,et al.Anti-markovnikov addition of both primary and secondary amines to terminal alkynes catalyzed by the TpRh(C2H4)2/PPh3system[J].J Am Chem Soc,2007,129(45):13792-13793.
[24]SHAFFER A R,SCHMIDT J.Palladium(Ⅱ) 3-iminophosphine complexes as intermolecular hydroamination for the formation of imines and enamines[J].Organometallics,2008,27(6):1259-1266.
[25]SAVARIN C G,BOICE G N,MURRY J A,et al.DirectN-acetyl enamines formation:lithium bromide mediated addition of methyllithium to nitriles[J].Org Lett,2006,8(18):3903-3906.
[26]CHUN Y S,KO Y O,SHIN H,et al.Tandem Blaise-alkenylation with unactivated alkynes:one-pot synthesis ofα-vinylatedβ-enaminoesters from nitriles[J].Org Lett,2009,11(15):3414-3417.
[27]CHUN Y S,RU K Y,KO Y O,et al.One-pot synthesis of 2-pyridonesviachemo- and regioselective tandem Blaise reaction of nitriles with propiolates[J].J Org Chem,2009,74(19):7556-7558.
[28]CHUN Y S,LEE K K,KO Y O,et al.The first chemoselective tandem acylation of the Blaise reaction intermediate:a novel method for the synthesis ofα-acyl-β-enamino ester,key intermediate for pyrazoles[J].Chem Commun,2008(41):5098-5100.
[29]KO Y O,CHUN Y S,PARK C L,et al.An effective and general method for the highly regioselective synthesis of 1-phenylpyrazoles fromβ-enaminoketoesters,tandem Blaise-acylation adducts[J].Org Biomol Chem,2009,7(6):1132-1136.
[30]LEE J H,CHOI B S,CHANG J H,et al.The decarboxylative Blaise reaction[J].J Org Chem,2007,72:10261-10263.
[31]THORWIRTH R,STOLLE A.Solvent-free synthesis of enamines from alkyl esters of propiolic or but-2-yne dicarboxylic acid in a ball mill[J].Synlett,2011(15):2200-2202.
基金项目:国家自然科学基金资助项目(21272170)
收稿日期:2016-03-20
作者简介:王艳红(1989-),女,山东菏泽人,硕士研究生,研究方向:Wittig烯化反应,E-mail:2013210123@tju.edu.cn;通讯作者:黄文华,副教授,E-mail:huangwh@tju.edu.cn。
doi:10.3969/j.issn.1672-5425.2016.07.001
中图分类号:O 623.7TQ 463.4
文献标识码:A
文章编号:1672-5425(2016)07-0001-06
Research Progress on Synthesis of Enamines
WANG Yan-hong,HUANG Wen-hua
(DepartmentofChemistry,TianjinUniversity,Tianjin300072,China)
Abstract:In this paper,the recent progress on synthesis methods of enamines was reviewed,including condensation method of amines with ketones or aldehydes,transition metal-catalyzed coupling reaction method,reduction method of ketoximes or amides and addition reaction method of alkyne or nitrile.Meanwhile,the advantages and disadvantages of each synthesis method were introduced,which provided a significant guidance for the choice of synthesis methods of enamines.
Keywords:condensation;transition metal-catalyzed coupling;reduction method;addition reaction;enamine
王艳红,黄文华.烯胺合成的研究进展[J].化学与生物工程,2016,33(7):1-6.
