SIRT1基因敲除对骨关节炎小鼠VEGF/AKT信号通路的影响
2016-08-07于斐曾晖雷鸣熊奡肖德明袁昊
于斐 曾晖 雷鸣 熊奡 肖德明 袁昊
北京大学深圳医院骨关节科,深圳 518036
骨关节炎(Osteoarthritis, OA) 是一种以关节软骨退变为特点,导致软骨基质降解、软骨细胞死亡和关节完整性破坏的慢性退行性病变,55岁以上人群的发病率为44%~70%[1]。目前,对于OA中软骨退变的具体机制尚未完全阐明,而对其基因水平的研究近年来备受关注。
沉默信息调节因子1(SIRT1)是一个含有高度保守催化核心序列的基因,依赖于烟酰胺腺嘌呤二核苷酸,调控许多下游基因的转录,从而起到抑制或激活下游基因、调节机体代谢、延长寿命和控制细胞分化、再生以及存活等多个方面的生物学效应[2]。其可以通过抑制凋亡、调控新陈代谢、维持氧化应力下线粒体的正常功能以及抑制炎症等多个方面来调控细胞的衰老[3]。VEGF/AKT通路在生物体中起到重要作用,该通路可以通过血管异常生成加强炎症的发生,并通过调节软骨细胞的存活和凋亡影响OA中关节软骨的退变过程。
本研究应用Cre-Lox系统构建软骨特异性敲除SIRT1基因(SIRT1 cKO)小鼠,并通过单侧膝关节前交叉韧带横断加内侧半月板切除术建立OA模型,观察SIRT1基因敲除后是否通过VEGF/AKT通路对OA关节软骨退变产生影响,探讨SIRT1基因通过VEGF/AKT通路调节OA关节软骨退变的具体机制。
1 材料与方法
1.1 材料
1.1.1实验动物:SIRT1co/co小鼠和Col2-CreERT2小鼠均购买自美国杰克逊实验室。实验用小鼠饲养于北京大学/香港科技大学医学中心深圳医院实验动物中心SPF级屏障区域的PVC鼠笼内,持续过滤通气,自由饮食,清洁饮水,每日保持12 h光照/黑暗循环。实验流程和小鼠处理均遵循实验动物管理规范条例。
1.1.2主要仪器:光学显微镜(德国Leica公司),高速冷冻离心机(德国SIGMA公司),高压灭菌器(日本Hirayama公司),PCR热循环仪(德国Eppendorf公司),凝胶成像系统Dolphin-DOC(美国Weahec公司),ABI Prism 7000型荧光定量PCR仪(美国ABI公司),包埋切片机(日本Sakura公司)。
1.1.3主要试剂:VEGF、AKT、SIRT1一抗(Abcam公司),生物素化兔抗山羊IgG、SABC-POD、DAB显色试剂盒(武汉博士德生物工程有限公司),他莫昔芬、玉米油(Sigma公司),盐酸赛拉嗪注射液(吉林省敦化市圣达动物药品有限公司),TRE-trizol(Invitrogen公司), PrimeScript II 1st Strand cDNA Synthesis Kit、SYBR Premix Ex Taq II(TaKaRa公司),Fast Green Solution、Safranin O Solution(Scytek公司)。
1.2 方法
1.2.1软骨特异性敲除SIRT1基因小鼠的繁育:将Col2-CreERT2小鼠与SIRT1co/co小鼠交配,获得Col2-CreERT2;SIRT1co/co小鼠(SIRT1+/+小鼠),15个月月龄Col2-CreERT2;SIRT1co/co小鼠腹腔注射Tamoxifen(10mg/ml,1mg/ml·g体重,每日1次,连续注射5d),即获得SIRT1 cKO小鼠(SIRT1-/-小鼠)。
1.2.2实验分组及膝关节骨关节炎模型的建立:Tamoxifen注射完毕7 d后,SIRT1+/+小鼠和SIRT1-/-小鼠行右侧膝关节前交叉韧带横断加内侧半月板切除术建立OA模型,SIRT1+/+小鼠左侧膝关节仅切开滑膜囊行假手术作为对照,手术后3个月建立膝关节OA模型。实验样本分为两组:SIRT1+/+小鼠骨关节炎模型组(A组,n=6):SIRT1基因未敲除,膝关节手术造成OA模型;SIRT1-/-小鼠骨关节炎模型组(B组,n=6):SIRT1基因敲除,膝关节手术造成OA模型。模型建立后颈椎脱臼法处死小鼠,留取双侧膝关节,10%福尔马林固定后石蜡包埋。
1.2.3荧光定量PCR反应鉴定软骨特异性敲除SIRT1基因小鼠的基因表型: 根据NCBI网站GeneBank查询目的基因SIRT1序列,引物合成使用Primer Premier 5.0软件,序列见表1。cDNA合成采用逆转录试剂盒。按照各目的基因退火温度选择三步法完成扩增,循环44次。根据熔解曲线判断扩增产物的特异性,以GAPDH为内参。反应参数参考TaKaRa SYBR Premix Ex Taq II (Perfect Real Time)试剂盒说明进行,运用Bio-Rad CFX96 Real Time PCR仪配套的Bio-Rad CFX Manager Software1.6数据分析软件,采用2^-ΔΔCt法开展数据分析。
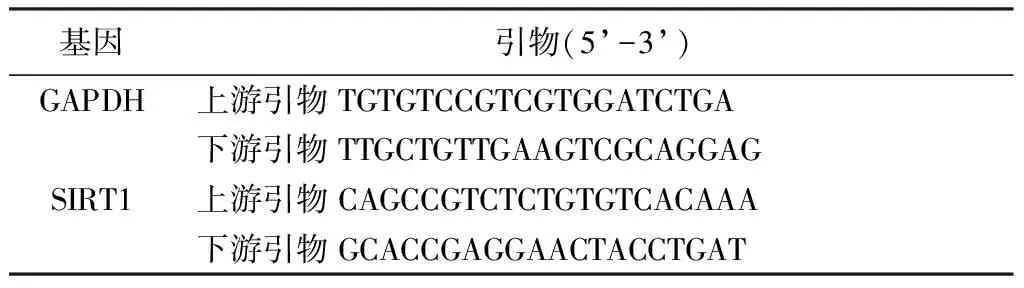
表1 荧光定量PCR反应鉴定软骨特异性敲除SIRT1基因小鼠的引物Table 1 SIRT1 gene primers
1.2.4软骨组织学检测:组织标本修切后10%多聚甲醛固定24 h,脱钙后石蜡包埋,连续切片,片厚 5 um,行苏木素-伊红(HE)、番红O-固绿双染色。按照文献[4]进行Mankin评分。
1.2.5免疫组化染色:烤片3 min;蒸馏水冲洗2次;抗原修复; PBS冲洗3次;滴加过氧化物酶阻断剂;PBS液冲洗3次;滴加非免疫性动物血清;滴加SIRT1、VEGF和AKT-抗;PBS液冲洗3次;滴加生物素标记的二抗;滴加SP溶液;滴加2滴新鲜配制的DAB溶液。
1.2.6免疫组化结果观察与判定:SIRT1、VEGF、AKT染色结果判定:经DAB显色,SIRT1、VEGF、AKT免疫组织化学以显微镜下染成棕黄色为阳性细胞;将染色强度分为4级:0级:阴性染色,1级:弱阳性染色,2级:中度阳性染色,3级:强阳性染色;再将每张切片中按所见阳性细胞范围分为5级:0级:无阳性细胞,1级:阳性细胞占l%~20%,2级:阳性细胞占21%~50%,3级:阳性细胞占51%~75%,4级:阳性细胞占76%~100%;每张切片染色积分以两者乘积表示。

2 结果
2.1 软骨特异性敲除SIRT1基因小鼠的鉴定
剪取A组小鼠和B组小鼠胸骨处关节软骨提取总RNA,并通过荧光定量 PCR反应鉴定小鼠基因表型,结果显示A组小鼠SIRT1 mRNA表达量为8.33467±1.800794,B组小鼠SIRT1 mRNA表达量为2.24417±1.316569,差异有统计学意义(P<0.01)(图1),证实SIRT1-/-小鼠关节软骨中SIRT1基因敲除成功。

图1 实验中所用小鼠SIRT1表达趋势及电泳图Fig.1 The expression of SIRT1 and electrophoresis of SIRT1 in the mice used in the experiment.注:**P<0.01;Note:**P<0.01.
2.2 软骨特异性敲除SIRT1基因小鼠膝关节软骨组织学观察
A组小鼠对照侧软骨组织层次清楚,关节表面软骨平整,软骨细胞正常,排列较整齐,分布均匀,染色均匀,番红O染色正常,潮线完整无血管通过,Mankin评分为2.6667±0.51640分;A组小鼠骨关节炎造模侧膝关节表面软骨毁损,形态不规则,发生糜烂,软骨变薄,软骨细胞分布不均匀,成簇排列,番红O染色不均匀,血管通过潮线,Mankin评分为7.3333±0.51640分,两侧Mankin评分差异有统计学意义(P<0.01),证实小鼠骨关节炎模型建立成功。B组与A组相比膝关节表面软骨毁损更严重,表面出现断裂,甚至发生软骨层缺失,软骨细胞数量减少,番红O失染,潮线前移且有血管通过,软骨退变更严重,B组Mankin评分为9.8333±1.94079分,与A组相比Mankin评分差异有统计学意义(P<0.05)(图2、3)。

图2 各组膝关节软骨组织学表现:HE染色,番红O固绿双染色,×100。A与D:SIRT1+/+小鼠假手术组膝关节表面软骨情况;B与E:SIRT1+/+小鼠骨关节炎模型组膝关节表面软骨情况;C与F:SIRT1-/-小鼠骨关节炎模型组膝关节表面软骨情况。Fig.2 The histology of the knee joint cartilage: HE staining and Safranin O-Fast Green staining, ×100.A and D: SIRT1+/+ mice in OA group; B and E: SIRT1+/+ mice in OA group; C and F: SIRT1-/- mice in OA group.

图3 SIRT1+/+小鼠骨关节炎模型组和SIRT1-/-小鼠骨关节炎模型组小鼠膝关节Mankin评分Fig.3 The Mankin’s score of SIRT1+/+ mice in OA group and SIRT1-/- mice in OA group.注:*P<0.05;Note:*P<0.05.
2.3 SIRT1、VEGF、AKT免疫组化染色
A、B两组膝关节软骨中SIRT1免疫组化染色积分分别为3.6667±1.21106、3.3333±1.96638;VEGF免疫组化染色积分分别为7.0000±1.54919、10.0000±2.44949;AKT免疫组化染色积分分别为3.1667±0.40825、1.3333±1.21106。SIRT1免疫组化结果中,B组与A组比较差异统计学意义不明显(P>0.05);VEGF免疫组化结果中,B组与A组比较差异有统计学意义(P<0.05);AKT免疫组化结果中,B组与A组比较差异有统计学意义(P<0.01)(图4、5)。

图4 各组膝关节软骨组织免疫组化,×400。A与B为SIRT1+/+小鼠骨关节炎模型组和SIRT1-/-小鼠骨关节炎模型组SIRT1表达情况;C与D为SIRT1+/+小鼠骨关节炎模型组和SIRT1-/-小鼠骨关节炎模型组VEGF表达情况;E与F为SIRT1+/+小鼠骨关节炎模型组和SIRT1-/-小鼠骨关节炎模型组AKT表达情况。Fig.4 The immunohistochemical images of the knee joint cartilage, ×400.A and B: The expression of SIRT1 in SIRT1+/+ mice in OA group and SIRT1-/- mice in OA group; C and D: The expression of VEGF in SIRT1+/+ mice in OA group and SIRT1-/- mice in OA group; E and F: The expression of AKT in SIRT1+/+ mice in OA group and SIRT1-/- mice in OA group.

图5 SIRT1+/+小鼠骨关节炎模型组和SIRT1-/-小鼠骨关节炎模型组两组SIRT1、AKT、VEGF在膝关节软骨中免疫组化染色积分。Fig.5 Immunohistochemical staining integral of SIRT1, AKT, and VEGF in the knee joint cartilage in SIRT1+/+ mice in OA group and SIRT1-/- mice in OA group.注:**P<0.01,*P<0.05;Note:**P<0.01,*P<0.05.
3 讨论
OA以关节软骨的退变磨损为主要特征,近年的研究表明长寿基因SIRT1在OA关节软骨退变中起重要作用,该基因与衰老关系密切,是多种退行性病变发生的根源。
SIRT1基因可通过调节通路中蛋白的乙酰化影响软骨细胞存活,使软骨区域糖蛋白减少,钙元素沉积加速,导致关节软骨退变[5]。OA发生时,软骨细胞中SIRT1的表达水平明显降低,抑制SIRT1表达可引起软骨细胞凋亡[6]。而凋亡不仅加速成骨细胞和血管进入,还伴有钙的积累和基质钙化,加速关节软骨病理性退变。因此,SIRT1基因在OA关节软骨的退变中起到重要作用。为了研究SIRT1基因的功能及作用机制,我们把软骨特异性敲除SIRT1基因小鼠,和与其具有相同品系、月龄和饲养条件的SIRT1基因未敲除小鼠作对照,最大限度减少其他因素干扰。而Mankin评分是对OA评价的一种评分标准,分数越高,OA程度越严重,软骨退变越明显,结合该评分可以说明骨关节炎的发生发展程度。为了模拟OA的好发年龄,我们使用15月龄小鼠进行实验[7]。
我们的研究发现,Tamoxifen诱导后小鼠的软骨组织中SIRT1基因敲除成功,当SIRT1基因敲除后,OA发生时小鼠的关节软骨退变程度更严重,Mankin评分与SIRT1+/+小鼠OA模型组相比升高更明显;SIRT1基因未敲除时,发生OA关节软骨退变的一侧膝关节中SIRT1的表达量显著下调,Mankin评分与SIRT1+/+小鼠假手术侧相比显著升高。这说明SIRT1基因敲除可以加重OA关节软骨的退变,并且实验中骨关节炎小鼠模型建立成功,当OA发生时,关节软骨中SIRT1表达水平明显下调。
VEGF/AKT信号通路作为一个与血管生成密切相关的通路[8],它在促进血管内皮生成、完整性维持及内皮细胞增殖分化方面起重要作用,同时它还可以影响细胞的衰老凋亡,对病理性退变产生作用。研究证实[9,10],VEGF可以激活AKT,共同影响新生血管中内皮细胞增值分化,加速新生血管的形成;激活内皮细胞中SIRT1表达后可引起细胞中AKT表达上调,减轻内皮细胞凋亡,血管生成速度减慢,并且用VEGF刺激时,血管内皮细胞的生成又恢复正常,血管生成加速,SIRT1过表达后AKT的磷酸化水平升高,其通过下调氧化应激抑制血管内皮的衰老。
在OA中,VEGF/AKT通路通过血管异常生成加强炎症发生,并通过调节软骨细胞的存活影响关节软骨退变。其中,VEGF诱导的血管生成分布在骨软骨连接处,该处的血管生成与软骨退化、疾病活动性相关[11]。在正常组织中,关节软骨和软骨下骨交界处的血管生成和抗血管生成处于平衡状态,而OA的滑膜炎症刺激了血管生成,诱导软骨发生退变,导致潮线前移、骨赘形成。随着软骨基质被血管侵蚀,VEGF与AKT影响软骨细胞导致钙化层内的细胞发生凋亡, 软骨基底被新生的骨组织取代。当OA关节部位骨赘生成时,该区不仅出现VEGF的表达上调[12]而且在下调的AKT协同作用下出现软骨细胞凋亡。也有研究表明[13,14],在OA中,软骨变性、软骨基质成分改变可引起VEGF和AKT作用,导致血管的生成,因炎症致敏的神经纤维又引起炎症,进一步刺激血管生成;增多的VEGF也引起基质金属蛋白酶等因子升高,分解软骨基质,破坏软骨组织,这些升高的因子又诱导VEGF的表达,形成恶性循环,加重OA的软骨退变。而AKT是一个比较保守的基因,可以调节能量的代谢,与细胞的寿命相联。该基因可能反式作用于VEGF的启动子,促进VEGF的表达分泌[15];AKT本身可以通过磷酸化下游基因促进软骨细胞的存活,负向调控p53抑制软骨细胞凋亡,并可以产生抗凋亡作用,AKT还可降低下游过氧化氢酶的表达,使细胞更易受到活性氧的影响发生衰老凋亡[16,17]。
那么在OA关节软骨的退变中,SIRT1与VEGF/AKT通路是否存在着关联?我们通过实验发现,SIRT1基因敲除与未敲除两组小鼠中,当OA发生时VEGF和AKT的表达量均发生变化,其中VEGF发生上调,AKT发生下调。而当SIRT1基因敲除后,OA发生时VEGF的上调与AKT的下调比SIRT1基因未敲除时更为显著。这可能是由于SIRT1基因敲除后VEGF/AKT通路被激活,上调的VEGF与下调的AKT协同作用引起钙化区软骨细胞向肥大表型分化进而促进软骨细胞发生去分化和细胞凋亡[13,16],而VEGF引起滑膜、关节软骨和软骨下骨连接处异常血管生成并侵入凋亡细胞所在位置[11],在引起神经源性炎症的同时导致软骨基质分解、软骨基底发生骨化并被新生骨组织取代,潮线前移的同时引起骨赘生成,而骨赘区软骨细胞也在VEGF和AKT的作用下出现凋亡[12,16],进而软骨组织发生病理性退变和钙化,骨赘形成、软骨基质成分的改变、软骨的退化又进一步导致VEGF分泌增加而AKT分泌减少,形成恶性循环,最终加重OA中软骨组织退变。
OA中关节软骨退变的具体机制至今尚未完全阐明,而特定基因如何通过信号通路影响关节软骨的变化近年来备受关注。我们在建立膝关节OA的小鼠模型上阐明了SIRT1基因敲除可能通过激活VEGF/AKT这一通路加重OA关节软骨的退化,因此SIRT1基因可能对骨关节炎起到保护作用,这对于研究骨关节炎的发病机制及治疗措施具有一定的意义。
