气相中Ti2+活化环己烷同面脱氢机理的研究
2016-08-02柳瑞瑞耿志远
柳瑞瑞 , 耿志远
(西北师范大学 化学与化工学院 , 甘肃 兰州 730070)
气相中Ti2+活化环己烷同面脱氢机理的研究
柳瑞瑞 , 耿志远
(西北师范大学 化学与化工学院 , 甘肃 兰州730070)
摘要:采用密度泛函理论(DFT)的B3LYP方法对2+活化环己烷的同面脱氢机理进行理论计算,分别得到反应中涉及到的驻点、优化构型及相关的构型参数,并简单绘制了反应势能图,从而对反应机理进行详细的分析。对环己烷与2+反应的同面脱氢机理进行研究,研究结果表明环己烷与2+的同面脱氢过程中三次脱氢机理相似,反应发生在混合势能面上,最终产物是二、四重态的混合物,且放热分别为54.85、28.74 kJ/mol。
关键词:脱氢机理 ; 环己烷 ;; DFT 理论研究
0引言
环己烷是一种非常重要的化学药品,在天然绿色能源中得到广泛利用,尤其以氢能为主[1-4]。在环己烷中,C—H键的活化是一个重要的能源过程,因为环己烷比其他的材料具有更好的储氢功能,例如压缩氢气、液体氢、氢储藏金属合金以及碳材料[5-9]。因此,更多的科学家对于研究环己烷脱氢有很大的兴趣。
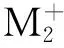

1计算方法
采用了密度泛函理论(DFT)的B3LYP方法,应用量子化学通用的计算软件Gaussian09程序,对于C、H原子,使用了6-31G(d,p)基组,对于Ti2+,采用LANL2DZ赝势基组,将所有中间体以及过渡态的结构进行了全优化;通过对各中间体的频率分析,说明反应物、中间体和产物都是稳定点,每个过渡态的频率只有一个虚频[14-15]。用内禀反应坐标(IRC)[16]验证了反应沿着虚频振动方式分别趋于反应物和产物,并且运用了Yoshizawa等[17]提出的内禀坐标垂直激发单点计算法确定不同自旋态下两个势能面的交叉点(crossing point, CP),即用任意自旋态内反应坐标点所对应的构型,对该构型在二、四重态下的单点能进行计算,从而得到其中一个自旋态势能面上的内禀反应坐标点在另外一个自旋态势能面上的投影曲线,进而找到一个自旋态势能面上的IRC曲线和另外一个自旋态势能面间的交叉点,采用Crossing 2004和GAMESS程序包,在交叉点的基础上再找到对应的MECP并计算相对应的SOC值。对部分重要的中间体,做了NBO分析[18]。
2结果与讨论
2.1反应物结构
图在二、四重态的分子轨道能级图


图2 反应在二、四重态下的同面脱氢势能图


图3 同面脱氢反应中二、四重态下第一分子的脱氢反应中各驻点几何构型
各驻点几何构型如图4所示。
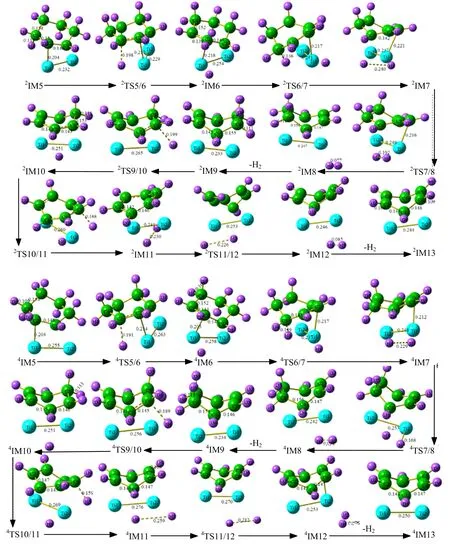
图4 同面脱氢反应中二、四重态下发生第二、三分子脱氢的各驻点的几何构型
2.3最低能量交叉点(MECP)与自旋—轨道耦合常数(SOC)

图5 2IM2→2TS1/2 (a),2TS5/6→2IM6 (b)过程中CP1、CP2和MECP1、MECP2的构型图
如图2所示,基态二重态反应物能量比四重态低21.17 kJ/mol,但是二重态的过渡态2TS1/2的能量比四重态高28.11 kJ/mol,这说明交叉点生在过渡态2TS1/2之前,我们应用上述提到的相关方法和程序在此处找到了CP1和MECP1如图5所示。同时也计算出了MECP1的自旋耦合常数(SOC)为398.12 cm-1,利用相同的方法,我们找到了CP2和MECP2,同时也计算出了MECP2的自旋耦合常数(SOC)为312.45 cm-1,其SOC值均大于100 cm-1,说明在MECP1、2处发生了有效的电子自旋翻转。
为了更好地理解电子组态和每个最低能量交叉点处的机理,我们进行了NBO分析,如图6所示。
图6中ψ(a)主要代表Ti2的3dxz轨道,这也是2MECP1的LUMO轨道和4MECP1的HOMO轨道,ψ(c)主要代表Ti2的3dxy轨道, 在MECP1中,Ti2的3dxy上的β电子翻转进入Ti2的3dxz轨道,在MECP1处,二重态转向四重态并降低了反应势垒。在图b中,ψ(a)主要代表Ti2的4s和3dz2轨道,这也是4MECP2的HOMO轨道和2MECP2的LUMO轨道,在MECP2处,ψ(c)主要代表Ti2的3dz2轨道,Ti2的杂化轨道上的α电子翻转进入Ti2的 轨道并完成了四重态向二重态转变的过程,前线分子轨道的分析说明多重态在MECP处的相互转化是可行的。
3结论
参考文献:
[1]Nouryzadeh H,Iranshahi D.Hydrogen and gasoline production through the coupling of fischer-tropsch synthesis and cyclohexane dehydrogenation in a thermally coupled membrane reactor[J].Petroleum Coal,2014,56:231-248.
[2]Markiewicz M,Zhang Y Q,smann A B,et al.Environmental and health impact assessment of Liquid Organic Hydrogen Carrier (LOHC) systems-challenges and preliminary results[J].Energy Environ.Sci.2015(8):1035-1045.
[3]刘凯,安越,徐国华,等.环己烷连续脱氢反应研究[J]. 西安交通大学学报. 2007,41(12):1495-1498.
[4]陈进富,蔡卫权,俞英.有机液态氢化物可逆储放氢技术的研究现状与展望[J].太阳能学报.2002,23(4): 528-532.
[5]Ciobica I M,Frechard F,Van Santen R A,et al. A DFT study of transition states for CH activation on the Ru (0001) surface[J].The Journal of Physical Chemistry B. 2000,104(14): 3364-3369.
[6]Strassner T, Muehlhofer M, Zeller A, et al. The counterion influence on the CH-activation of methane by palladium (II) biscarbene complexes-structures, reactivity and DFT calculations[J]. Journal of organometallic chemistry. 2004, 689(8): 1418-1424.
[7]Ciobica I M,Van Santen R A.A DFT study of CHxchemisorption and transition states for CH activation on the Ru (1120) surface[J]. The Journal of Physical Chemistry B. 2002, 106(24): 6200-6205.
[8]SchrÖder D,Schwarz H.Gas-phase activation of methane by ligated transition-metal cations[J].P. Natl. Acad. Sci. 2008,105:18114-18119.
[9]Du J,Wu J,Guo T,et al.Catalytic performance of Mo2C supported on onion-like carbon for dehydrogenation of cyclohexane[J].RSC Adv,2014,4:53950-53953.
[10]Seemeyer K,Schroder D,Kempf M, et al.Face selectivity of the C—H bond activation of cyclohexane by the “bare” first-row transition-metal cations Sc+-Zn+[J].Organometallics.1995,14:4465-4470.
[12]Schlangen M,Schwarz H.Probing elementary steps of nickel-mediated bond activation in gas-phase reactions:ligand- and cluster-size effects[J].J Catal,2011,284:126-137.

[14]Becke A D. Density‐functional thermochemistry. III. The role of exact exchange[J]. J Chem Phys,1993,98(7): 5648-5652.
[15]Wadt W R, Hay P J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi[J].J Chem Phys,1985, 82(1): 284-298.
[16]Fukui K.The path of chemical reactions - the IRC approach[J]. Acc Chem Res,1981, 14(12): 363-368.
[17]Yoshizawa K, Shiota Y, Yamabe T. Intrinsic reaction coordinate analysis of the conversion of methane to methanol by an iron-oxo species: a study of crossing seams of potential energy surfaces[J].J Chem Phys,1999, 111(2): 538-545.
[18]Harvey J N, Aschi M, Schwarz H,et al. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces[J].Theor Chem Acc,1998, 99(2): 95-99.
[19]Jiang R W,But P P -H,Ma S -C,et al.Structure and antiviral properties of macrocaesalmin, a novel cassane furanoditerpenoid lactone from the seeds of Caesalpinia minax Hance[J].Tetrahedron Lett,2002,43:2415-2418.
收稿日期:2016-03-25
作者简介:柳瑞瑞(1990-),女,硕士,研究方向是计算量子化学,电话:18294435134。
中图分类号:O621.25+4.3
文献标识码:A
文章编号:1003-3467(2016)05-0033-06
Study on Same-face Dehydrogenation Mechanism of C6H12Activated by2+in Gas Phase
LIU Ruirui , GENG Zhiyuan
( School of Chemical and Chemical Engineering , Northwest Normal University , Lanzhou730070 , China)
Abstract:The mechanism of catalyzed cyclohexane dehydrogenation has been studied by the B3LYP level of density functional theory (DFT).The stagnation point, optimizing configuration and related parameters are obtained,and the reaction potential energy is drawn,and the reaction mechanism is analyzed in detail.The same-face dehydrogenation mechanism of cyclohexane catalyzed by is studied,the results show that every molecular dehydrogenation mechanism of the same-face dehydrogenation is similar,and the reaction proceed in mixed potential energy surface,final product is mixtures of doublet,quartet state,which is exothermic by 54.85 kJ/mol for the doublet and 28.74 kJ/mol for the quartet states.
Key words:dehydrogenation mechanism ; cyclohexane ; ; DFT theoretical study
