A型流感病毒基质蛋白M1的二聚化机制
2016-07-28张一堪刘建平
张一堪,曹 密,刘建平,黄 强,3
(1. 复旦大学 生命科学学院 遗传工程国家重点实验室,上海 200438;2. 国家蛋白质科学中心(上海),上海 201203; 3. 上海生物制造技术协同创新中心,上海 200237)
A型流感病毒基质蛋白M1的二聚化机制
张一堪1,曹密2,刘建平1,黄强1,3
(1. 复旦大学 生命科学学院 遗传工程国家重点实验室,上海 200438;2. 国家蛋白质科学中心(上海),上海 201203; 3. 上海生物制造技术协同创新中心,上海 200237)
摘要:流感病毒基质蛋白1(matrix protein 1, M1)位于病毒包膜之下,形成一层壳状结构 (衣壳),可与病毒的血凝素、神经氨酸酶、包膜和病毒遗传物质发生相互作用,在病毒的组装与复制过程中起着关键作用.不过,除N 端有晶体结构外,全长M1蛋白的结构尚未解析.因此,人们对全长M1蛋白如何二聚化、然后多聚化形成病毒衣壳的过程知之甚少.为了解M1蛋白的二聚化机制,首先从M1的N 端片段晶体结构出发,用蛋白质结构预测方法获得M1的全长结构模型;其次,对可能的M1二聚体进行分子动力学模拟,分析其二聚化界面的氨基酸,由此发现了一种通过M1蛋白C端片段结合而形成的二聚体;最后,为验证模拟结果,用电镜三维重构方法分析了全长M1二聚体的构象,并提出了M1多聚化的机理模型.
关键词:A型流感病毒; 基质蛋白M1; 二聚化; 分子建模; 单颗粒三维重构
流感病毒是一种造成人类和动物患流行性感冒的RNA病毒,可引发急性上呼吸道感染、肺炎或心肺衰竭等[1].病毒基质是存在于病毒囊膜下,由基质蛋白1(matrix protein 1, M1)聚合形成的壳状结构[2],并把vRNP粒子包裹到壳内[3-4].M1既可以与病毒内部的遗传物质,又可以与膜蛋白和脂双层相互作用[3],在病毒囊膜形成过程中起到组织者的作用[5],因此在流感病毒的复制过程中扮演重要角色[6].M1的突变研究表明: 当把M1缺失的突变毒株感染宿主时,发现病毒丧失了侵染细胞的活力[7];当M1的一些氨基酸发生突变时,病毒的形状发生了改变,如变成球形、多角形、棒状,甚至会形成纤维状[8-9];M1组装时螺旋转角的不同也会使病毒粒子的形态呈现从球形到纤维状的变化[10].所以,在病毒复制和组装过程中,M1蛋白对病毒粒子的形态和功能有重要影响.因此,详细了解M1蛋白聚合组装的分子机理,揭示病毒的组装机制,可以为流感的预防和治疗提供新思路,意义重大.特别地,二聚化是M1蛋白多聚化过程的起始步骤[11],研究清楚M1二聚化的机制是解决该科学问题的前提与关键.
目前,对M1蛋白二聚化的结构分析还基本局限于M1的N 端片段(1~158aa).一个主要的原因是用晶体学方法很难获得全长M1蛋白 (1~252aa) 的结构,可能是因为: M1的C 端片段(159~252aa)结构柔性较大,且部分区段(159~164aa)易受蛋白酶水解而发生降解[12-13]等.1997年,Sha和Luo等[14]解析了M1蛋白N 端片段二聚体的晶体结构,发现N 端单体结构由约9个α螺旋构成,命名为M区域(螺旋H6~H9)和N区域(螺旋H1~H4),二聚化界面由平行的两个N端H6螺旋之间的疏水氨基酸作用 (Pro 90,Met 93,Val 97) 和N区域的氢键网络所维持,称之为平行式二聚化结构.2001年,Arzt等[12]解析出了N端二聚体的另外一种晶体结构[12],由两个单体堆叠形成,称之为堆叠式二聚化结构,二聚化界面面积约为1100 Å2.另外,小角散射的研究也表明,M1的结构N 端与C 端在溶液中呈现出了不同的运动状态: N 端片段的结构紧凑、稳定,而C 端片段结构伸展,且部分区域柔性较大[15].这些工作对M1的N端结构及其二聚化模式有了一定研究,但全长M1蛋白的结构和二聚化机制的研究还十分缺乏,因而也限制了人们对全长M1蛋白多聚化机理的理解.例如,虽然人们在N端片段二聚体晶体结构的基础上提出了初步的M1多聚化的机理模型,但是蛋白C端片段在该模型中的具体位置不清楚[16].
针对上述问题,为了解全长M1蛋白二聚化的机制,本工作从构建全长M1二聚体模型入手,用分子动力学(Molecular Dynamics, MD) 模拟方法研究全长M1蛋白可能的二聚化模式,揭示全长M1二聚体的结构,并利用负染电镜方法采集M1蛋白的图像,用单颗粒三维重构方法获得M1聚合体的构象,与MD模拟结果进行了比对分析.根据所得的全长M1二聚体模型,我们提出了全长M1多聚化的机理模型,为进一步揭示流感病毒自组装的过程提供基础.
1计算方法
1.1全长M1二聚体模型的构建
本研究选择了H1N1病毒(A/Puerto Rico/8/1934, H1N1)和H9N2病毒(A/chicken/Jiangsu/7/2002, H9N2)的M1蛋白作为研究对象.为建立全长M1蛋白的二聚体结构模型,首先以M1蛋白 N端片段晶体结构为模板,用同源模建程序MODELLER[17]预测全长结构,然后把全长结构分别与N端片段二聚体晶体结构[18]的两个单体进行结构比对,获得与N端片段类似的全长M1的二聚体模型,作为后续MD模拟的初始构象.
1.2MD模拟系统构建与模拟
本研究使用NAMD程序[19]进行MD模拟研究.以上述方法构建的M1二聚体模型为初始构象,用CHARMM力场描述系统原子的各种参数,水分子的模型为TIP3P,用VMD[4]程序按标准全原子显式溶剂方式构建模拟系统: 把M1二聚体置于立方盒子中央,蛋白质表面离盒子边界至少15Å,然后把水分子加入盒子中并加入相应数目的离子(Na+与Cl-),使体系的离子浓度值为0.150mol/L,并保持模拟系统电中性[20].
MD模拟计算步骤为: 首先对体系进行能量最小化,以消除系统内不合理的原子碰撞;接着,对体系进行时间尺度为250ns的基于NPT系统的模拟,体系温度设为300K,压强为1.01325×105Pa,积分步长为2fs,长程静电相互作用使用Particle Mesh Ewald (PME)求和方法计算.模拟中,每隔10ps保存一次体系原子的坐标与速度,以用于后续分析.NAMD输出的dcd轨迹文件是二进制格式,记录了模拟系统原子在各个时刻的坐标及速度.用可视化软件(如VMD)可以直观地观察模拟过程中蛋白质的构象变化及了解蛋白质内部分子振动状态的变化等[21].
1.3MD 模拟轨迹分析
1.3.1M1二聚体构象聚类分析
MD构象的聚类分析可以了解M1蛋白在模拟过程中的优势构象.我们使用分子模拟软件GROMACS(5.0.1版)中基于均方根偏差(Root-Mean-Square Deviation, RMSD)的gmx_cluster模块对M1二聚体进行聚类分析[22].分析时,从4个模拟体系50~250ns的模拟轨迹等时间间隔选取2000个构象进行聚类计算,聚类分析用的RMSD截断值为2.5Å.
1.3.2M1二聚化界面氨基酸分析
蛋白质二聚化界面是由两个单体特定位置的氨基酸相互作用而产生,这些特定的氨基酸称之为界面氨基酸.本研究采用MDcons程序[23]分析界面氨基酸.MDcons通过计算轨迹中不同单体间界面氨基酸残基的接触次数,来表征界面残基的保守性.两界面残基接触次数越多,表明此氨基酸在界面上的保守性就越高,反之越低.在计算时,氨基酸-氨基酸间相互接触的判据是它们的最近非氢重原子距离小于5Å[23].
2实验方法
2.1H9N2 M1蛋白的表达与纯化
将H9N2的M1蛋白基因(由上海生物制品研究所陈则研究员、上海计划生育研究所徐万祥研究员提供)克隆于原核表达载体pET-28a,转化E.coliBL21(DE3)构建表达M1蛋白的大肠杆菌工程菌.37℃ 培养工程菌至OD600值为0.4~0.6,降低温度至30℃,加入IPTG至终浓度为0.5mmol/L继续培养1h 后,收集菌体,超声破碎、离心后将上清液经镍柱吸附层析,获得M1蛋白质粗品.随后采用Sephacryl S200分子筛柱层析,对M1蛋白初始样品进一步精细纯化,收集M1蛋白组分,并浓缩至浓度为0.2mg/mL.样品存于-80℃,以用于后续电镜分析实验.
2.2负染电镜样品制备与图像采集
负染电镜数据采集在国家蛋白质科学中心(上海)的电镜分析系统上完成.取-80℃的M1蛋白样品冻融后,用pH 7.4缓冲液(20mmol/LTris,150mmol/L NaCl)分别稀释至2.5,5,10,20,30μg/mL,100μL冰上放置,4℃过夜.负染时,取300目(≈8.47μm)铜网的碳膜,PELCO EasiGlow辉光放电系统辉光放电10s,用镊子将铜网夹起,悬空放置,加稀释后的蛋白质样品10μL,孵育60s.然后,迅速用滤纸片接触铜网边缘将多余的溶液吸去;加10μL 1%(质量浓度)甲酸双氧铀溶液染液,孵育60s;随后,迅速吸去多余的染液,红外灯下15min后,将样品通过样品杆置入200kV FEI TECNAI TF20 场发射透射电子显微镜,观察并手动采集图像.
2.3单颗粒三维重构分析
单颗粒三维重构分析与图像显示主要使用软件EMAN[24]和Chimera[25]来进行.利用电镜图像进行单颗粒三维重构的一般流程请参考有关文献[26-27].
3结果
3.1全长M1二聚体的模型
以M1蛋白 N端晶体结构为模板,用同源模建方法构建了H1N1(A/Puerto Rico/8/1934, H1N1)和H9N2(A/chicken/Jiangsu/7/2002, H9N2) 病毒的M1二聚体的结构模型,作为后续MD模拟的初始构象,如图1所示.其中,根据平行式N端二聚体结构(PDB ID: 1aa7) 构建的H1N1全长M1二聚体模型称之为1aa7d,根据堆叠式N端二聚体结构 (PDB ID: 1ea3) 构建的全长 M1二聚体模型称为1ea3d;H9N2病毒对应的全长M1二聚体模型则分别称为: 1aa7m 和1ea3m.由于H1N1与H9N2的M1序列的几乎类似,个别氨基酸的差异对两个M1二聚体的初始构象影响很小.
3.2氨基酸残基的波动分析
为考察模拟体系是否达到平衡,以Cα原子RMSD随模拟时间的变化作为指标.以0时刻的构象为参考构象,4个模拟系统的M1的RMSD变化情况如图2所示.由图可见,在MD模拟进行到50ns后,4个体系都基本趋于稳定,说明模拟达到了平衡.因此,选取这4个模拟体系的50~250ns的轨迹进行后续分析.
蛋白质氨基酸残基的均方根涨落(Root Mean Square Fluctuation, RMSF)是残基位置与其平均位置的偏移量的平方进行时间平均后的开方值,反映了残基的自由运动幅度,即所谓的结构柔性.从图3可以看出,4个体系RMSF 值变化趋势一致,都是C 端片段原子位置波动性要远远高于N 端片段.可见,M1蛋白的C端片段结构柔性要高于N 端片段.
3.3M1二聚体在模拟中的优势构象分析
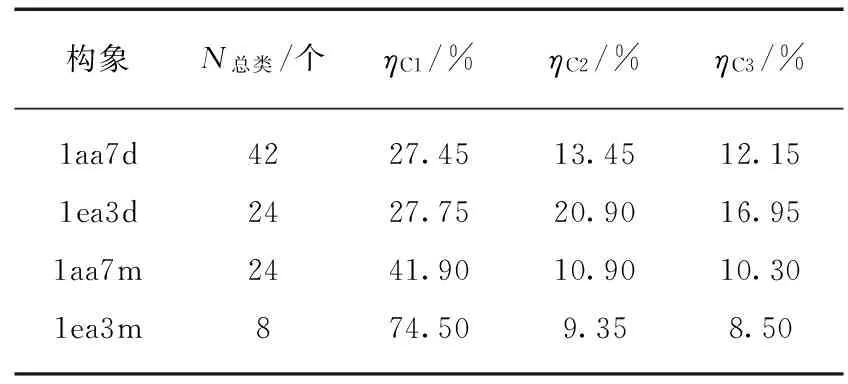
表1 4个模拟体系的MD构象的聚类结果
为获得全长M1二聚体在溶液中的优势构象,我们用GROMACS中的gmx_cluster模块对MD轨迹构象进行聚类分析,各体系中排序前三位的MD优势构象的比例(η)见表1,其聚类中心构象(即代表构象)见图4.由表1可知,每个体系中前三类构象占到全部MD构象的50% ,甚至以上.因此,我们选取前三个聚类的代表构象进行分析.另外,表1的数据说明,同一M1序列的平行式构象(1aa7d,1aa7m)构象聚类比堆叠式构象(1ea3d,1ea3m)多,表明在溶液中平行式构象的多态性高于堆叠式构象.这也说明了堆叠式构象在溶液中有更高的稳定性.从二聚体的空间构象也不难看出这一点,堆叠式构象的紧密度比平行式高,因此分子运动时所受的空间位阻作用比平行式大.
图4(见第392页)中的C1、C2 和C3 分别表示4个模拟体系中的比例最高的前三类聚类代表构象.由图可见,两种来源的M1结构的最稳定构象(C1)几乎没有差别,并且其N端结构都较稳定.另外,在两种平行式结构(1aa7d,1aa7m)中,我们发现其C端片段有向两边分散的趋势,如在(a)中的C1和C3的这种分散趋势较明显,再次表明C端片段的柔性高于N端片段.在堆叠式结构(1ea3d,1ea3m)中,由于空间位阻效应,这种趋势不及平行式结构明显具体表现在: 堆叠式结构中只有左上方的那条链对应的C端结构柔性较大,如在(c)中的C1和C3,而另外一条链(位于右下方)对应的C端结构在模拟中则较为稳定.另外,M1 蛋白是一种富α螺旋蛋白,二级结构约90% 由α螺旋组成.从聚类结果来看,α螺旋在聚类结果中占有较高的比例,说明全长M1预测结果的准确性.通过聚类分析,我们获得了M1二聚体在溶液中的优势构象,以下的分析将以这些结果为基础,如界面氨基酸分析所用构象为最大类的代表构象(即C1);电镜三维重构分析进行结构比较时用的也是该代表构象.
3.4全长M1二聚体界面氨基酸分析
用MDcons程序分析,我们获得了全长M1二聚体界面氨基酸的分布情况,如图5所示.对于平行式结构,相互作用界面氨基酸都分布在N端,与Sha和Luo[14]的结果一致,为极性氨基酸或疏水性氨基酸.这些氨基酸较容易形成α螺旋,因为N 端的氨基酸可与包膜相互作用,有比较多的疏水性氨基酸.在堆叠式M1二聚体结构,界面氨基酸在C端和N端都有分布.由M1蛋白组装的特性,我们推断平行式M1二聚体结构中也有部分界面氨基酸分布于C 端.于是,我们利用VMD通过设定周期性边界条件,观察4个模拟体系中多个二聚体的运动趋势,发现了新的一种M1二聚化的方式,即平行式构象用有C 端氨基酸组成界面的二聚化方式,其二聚化界面氨基酸情况如图5(e)所示.二聚化界面主要依赖于酸碱性氨基酸的相互作用来维持.我们把这种新发现的全长M1二聚体命名为1aa7c.
3.5H9N2 M1二聚体的电镜观测与分析
为获得用于电镜分析的蛋白质样品,用SDS-PAGE 电泳鉴定所纯化的H9N2 M1蛋白,结果如图6所示.由图可见,A280曲线有4个明显的峰,样品区分度明显.其中: 第一峰是外水体积;第二峰是M1的多聚体;第三个峰为主峰即二聚体;最后一个峰是易降解条带.
为获得M1二聚体形成的信息及其多聚化情况,我们使用200kV FEITECNAI TF20 场发射透射电镜下共拍得高清粒子图片345张,图片放大倍数为105倍.从中选取了有代表性的高清图像,如图7(a)所示.图中的单个颗粒为M1蛋白,中间有一条极淡的黑色的痕迹的颗粒为蛋白质二聚体,从图中可以观察到不同取向的二聚体.另外,从图中还可以看到体积较大的M1的多聚体.在本工作中,我们主要通过手动观察的方式选取电镜图像上的二聚体颗粒进行三维重构.图7(b)为二聚体颗粒的2D平均图及对应的结构模型图.M1二聚体的分子量约为53kD,用PyMOL测得3种构象1ea3d(1ea3m),1aa7d(1aa7m),1aa7c的三维尺寸分别为40Å×50Å×50Å,50Å×30Å×100Å,50Å×30Å×120Å.图7中显示的堆叠的粒子大多都呈长方体或立方体,其中间有淡淡的暗线,形状与下图的M1二聚体构象有明显的对应关系.在进行三维重构建模时共迭代8次,结构精修时迭代3次,所得密度图如图7(c)所示,分辨率约为14 Å.由图可见,三维重构密度图的中心位置与MD模拟中的堆叠式二聚体结构大致相似,其尺寸与同源模建的结构类似,由此我们推测在溶液中堆叠式二聚体构象较多.因此,通过负染电镜观察,MD模拟所获得的全长M1二聚体模型得到了初步验证.结合MD模拟的结果,我们提出了如图8所示的M1多聚化的模型.在此模型中,多聚化主要由计算分析所得的3个二聚化界面来驱动,即堆叠式二聚化界面(N端-C端),两种平行式二聚化界面(N端-N端和C端-C端).
4讨论
虽然M1蛋白N 端片段的二聚体结构已有报道[12,14],但由于M1蛋白C端片段易降解等原因,全长M1晶体结构还没有被解析.因此,在本研究中,我们采用理论计算和负染电镜相结合的手段对其二聚化的模式进行研究.报道中M1蛋白N端片段晶体结构的二聚化模式主要有平行式和堆叠式两种,故MD模拟时我们选择这两种模式作为初始模型.MD模拟的结果表明,全长M1的C 端片段的RMSF要高于N 端片段,这说明C端片段的结构柔性要高于N端,这也在一定程度上解释了全长晶体结构难以获得的原因.还有,通过分析了全长M1二聚体界面氨基酸的接触频率,了解二聚体界面的基本信息.在堆叠式结构中,二聚化界面氨基酸在N端与C端片段都有分布,其中,N端界面氨基酸与晶体学数据一致;对于平行式(N端-N端)全长M1二聚体来说,界面氨基酸多存在于77~134aa之间,与晶体结构的结果也保持一致.特别地,当对平行式二聚体结构进行进一步分析时,发现了另一个二聚化界面,其界面氨基酸主要由C 端片段氨基酸组成,因而获得了第三种M1全长二聚体的结合方式,如图5(e)所示.
另一方面,通过负染电镜方法分析了M1全长二聚体的真实构象,发现了与此模型一致的多聚体,如图7(a)右上方白色箭头部分.因此,综合了理论计算与实验结果而提出的多聚化模型(图8)与天然状态下M1的自组装是基本相符的.
综上所述,本文工作揭示了全长M1蛋白二聚化的3种可能模式,并由此提出了M1蛋白的多聚化模型,为理解M1聚合形成流感病毒衣壳的机理提供了有价值的结构信息,也为进一步了解流感病毒的组装过程提供了重要基础.
参考文献:
[1]陆德源主编.医学微生物学[M].北京: 人民卫生出版社,2001.
[2]RUIGROK R W, CRÉPIN T, HART D J,etal. Towards an atomic resolution understanding of the influenza virus replication machinery [J].CurrOpinStructBiol, 2010,20(1): 104-113.
[3]NAYAK D P, BALOGUN R A, YAMADA H,etal. Influenza virus morphogenesis and budding [J].VirusRes, 2009,143(2): 147-161.
[4]MURTI K G, WEBSTER R G, JONES I M. Localization of RNA polymerases on influenza viral ribonucleoproteins by immunogold labeling [J].Virology, 1988,164(2): 562-566.
[5]KRETZSCHMAR E, BUI M, ROSE J K. Membrane association of influenza virus matrix protein does not require specific hydrophobic domains or the viral glycoproteins [J].Virology, 1996,220(1): 37-45.
[6]ELTON D, DIGARD P, TILEY L,etal. Structure and function of the influenza virus RNP [M]∥ Kawaoka Y. Influenza virology: Current topics. Wymondham, United Kingdom: Caister Academic Press, 2006: 1-36.
[7]NAYAK D P, HUI E K W, BARMAN S. Assembly and budding of influenza virus [J].VirusRes, 2004,106(2): 147-165.
[8]BURLEIGH L M, CALDER L J, SKEHEL J J,etal. Influenza A viruses with mutations in the M1 helix six domain display a wide variety of morphological phenotypes [J].JVirol, 2005,79(2): 1262-1270.
[9]ROBERTS P C, LAMB R A, COMPANS R W. The M1 and M2 proteins of influenza A virus are important determinants in filamentous particle formation [J].Virology, 1998,240(1): 127-137.
[10]CALDER L J, WASILEWSKI S, BERRIMAN J A,etal. Structural organization of a filamentous influenza A virus [J].ProcNatlAcadSci, 2010,107(23): 10685-10690.
[11]ARAD G, LEVY R, NASIE I,etal. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock [J].PLoSBiol, 2011,9(9): e1001149.
[12]ARZT S, BAUDIN F, BARGE A,etal. Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer [J].Virology, 2001,279(2): 439-446.
[13]LUO B S M. Crystallization and preliminary X-ray crystallographic studies of type A influenza virus matrix protein M1 [J].ActaCrystallogrSectDBiolCrystallogr, 1997,53(4): 458-460.
[14]SHA B, LUO M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1 [J].NatStructMolBiol, 1997,4(3): 239-244.
[15]SHTYKOVA E V, BARATOVA L A, FEDOROVA N V,etal. Structural analysis of influenza A virus matrix protein M1 and its self-assemblies at low pH [J].PLoSOne, 2013,8(12): e82431.
[16]SAFO M K, MUSAYEV F N, MOSIER P D,etal. Crystal structures of influenza A virus matrix protein M1: Variations on a theme [J].PLoSOne, 2014,9(10): e109510.
[17]ESWAR N,WEBB B,MARTI-RENOM M A,etal. Comparative protein structure modeling using modeller [J/OL].CurrProtocinBioinform, 2006,5(6): 1-30.
[18]SEELIGER D, de GROOT B L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina [J].JComput-AidedMolDes, 2010,24(5): 417-422.
[19]JUNG J, MORI T, KOBAYASHI C,etal. GENESIS: A hybrid-parallel and multiscale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations [J].WIREsComputMolSci, 2015. doi: 10.1002/wcms.1220.
[20]HUMPHREY W, DALKE A, SCHULTEN K. VMD: Visual molecular dynamics [J].JMolGraphics, 1996,14(1): 33-38.
[21]LEACH A R. Molecular modelling: Principles and applications [M]. Berlin: Pearson Education, 2001.
[22]LINDAHL E, HESS B, VAN DER SPOEL D. GROMACS 3.0: A package for molecular simulation and trajectory analysis [J].JMolModel, 2001,7(8): 306-317.
[23]ABDEL-AZEIM S, CHERMAK E, VANGONE A,etal. MDcons: Intermolecular contact maps as a tool to analyze the interface of protein complexes from molecular dynamics trajectories [J].BMCBioinform, 2014,15(8): 1187-1198.
[24]MURRAY S, GALAZ-MONTOYA J, TANG G,etal. EMAN2.1-a new generation of software for validated single particle analysis and single particle tomography [J].MicroscMicroanal, 2014,20(S3): 832-833.
[25]PETTERSEN E F, GODDARD T D, HUANG C C,etal. UCSF Chimera-a visualization system for exploratory research and analysis [J].JComputChem, 2004,25(13): 1605-1612.
[26]张磊.脂蛋白结构与功能的透射电子显微镜研究 [D].西安: 西安交通大学,2010.
[27]FRANK J, RADERMACHER M, PENCZEK P,etal. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields [J].JStructBiol, 1996,116(1): 190-199.
[28]MARTI-RENOM M A, STUART A C, FISER A,etal. Comparative protein structure modeling of genes and genomes [J].AnnuRevBiophysBiomolStruct, 2000,29(1): 291.
文章编号:0427-7104(2016)03-0388-08
收稿日期:2015-07-20
基金项目:国家自然科学基金(91430112),上海市自然科学基金(13ZR1402400)
作者简介:张一堪(1988—),男,硕士研究生;黄强,男,教授,通讯联系人,E-mail: huangqiang@fudan.edu.cn.
中图分类号:Q 71
文献标志码:A
Dimerization Mechanisms of Influenza A Virus Matrix Protein M1
ZHANG Yikan1, CAO Mi2, LIU Jianping1, HUANG Qiang1,3
(1.StateKeyLaboratoryofGeneticEngineering,SchoolofLifeSciences,FudanUniversity,Shanghai200438,China;2.NationalCenterforProteinScienceatShanghai,Shanghai201203,China;3.ShanghaiCollaborativeInnovationCenterforBiomanufacturingTechnology,Shanghai200237,China)
Abstract:Influenza A virus matrix protein 1(M1) forms the viral capsid under the viral membrane,and interacts with hemagglutinin(HA),neuraminidase(NA),membrane and RNA simultaneously. However,crystal structures are only available for the N-terminal segment of M1. Little is known about the dimerization of the full-length M1 proteins that is required for the polymerization to form the capsid. To understand the dimerization mechanism of M1,here we used homology modeling to predict the structure of the full-length M1 on the basis of the N-terminal structure. Then,we carried out molecular dynamics(MD) simulation for the possible M1 dimers,and analyzed the amino acids of the dimerization interfaces. The results of MD simulation indicated there is a new dimerization interface formed by the interactions of two M1 C-terminal segments. To verify the results,we analyzed the M1 dimers by the single-particle 3D reconstruction of electron microscopy,and thereby presented a mechanistic model for the M1 polymerization.
Keywords:influenza A virus; matrix protein M1; dimerization; molecular modeling; single-particle 3D reconstruction
