高通量测序技术分析天然肠衣香肠的菌群组成
2016-07-27滕安国齐晓娜张芹王稳航天津科技大学食品工程与生物技术学院天津300457
滕安国,齐晓娜,张芹,王稳航(天津科技大学食品工程与生物技术学院,天津300457)
高通量测序技术分析天然肠衣香肠的菌群组成
滕安国,齐晓娜,张芹*,王稳航
(天津科技大学食品工程与生物技术学院,天津300457)
摘要:以贮藏至不同时期的天然肠衣香肠为研究对象,采用高通量测序技术,研究正常香肠、发黏香肠,以及腌渍天然肠衣的菌群组成。研究发现:贮藏至不同时期的香肠及肠衣之间,微生物多样性及丰度均有较大差异,正常香肠和肠衣微生物多样性及丰度均较高,发黏香肠微生物多样性较低,Lactobacillus和Leuconostoc为其优势菌群,在种的水平上分类,则是Lactobacillus_paraplantarum、Leuconostoc_mesenteroides_subsp_mesenteroides_J18为优势菌群。
关键词:天然肠衣香肠;微生物多样性;高通量测序技术
随着社会生活节奏加快,香肠需求巨大,特别是天然肠衣的乳化香肠作为烤肠、早餐肠等,出现在各类饮食场所。然而,天然肠衣的香肠货架期短,特别是在夏季,一般在3 d以内,否则极容易出现发黏、发酸等腐败现象。除香肠外,其它类型的加工肉制品在贮藏时,亦有类似情况发生。香肠腐败给生产企业带来极大的困扰,由于货架期短,生产企业无法规模化备货及长距离运输,严重影响经济效益和企业发展。处理此类棘手问题成为当前香肠生产企业面临的共性问题,目前此方面研究报道较少。本文以此为出发点,研究贮藏至不同时期的香肠菌群组成状况及丰度,为香肠生产企业有针对性的解决香肠腐败问题,延长产品货架期提供理论参考。
1 材料与方法
1.1材料
1.1.1材料
香肠采购自天津某肉制品生产企业,分别取喷淋抗菌剂的正常香肠(A)、未喷淋抗菌剂的正常香肠(B)及发黏香肠(C)和肠衣(D)。
1.1.2主要试剂和仪器
DNA提取试剂盒:DNA Tissue Ki(tQIAGEN,Germany)。PCR仪:ABI GeneAmpR9700型。
1.2方法
1.2.1香肠样品微生物基因组DNA提取与PCR扩增
取不同香肠各3根,参照Miambi等(2003)的方法[1],无菌条件下取各香肠的肠衣部分,并取生产香肠使用的腌渍肠衣。以上样品均截取10 g剪碎,放于50 mL生理盐水中,摇床振摇30 min,于4℃,4 000 r/min,离心10 min,取上清液20 mL于10 000 r/min,4℃条件下离心20 min,取沉淀于1.5 mL离心管,参照DNA Tissue Kit(QIAGEN,Germany)试剂盒说明书提取总的细菌DNA,溶于TE缓冲液,1.0%琼脂糖凝胶电泳检测DNA完整性,-20℃条件贮藏备用。
采用引物27F 5'-AGAGTTTGATCCTGGCTCAG-3',533R 5'-TTACCGCGGCTGCTGGCAC-3',扩增细菌16SrRNA的V1-V3区。PCR反应体系为:5× FastPfu缓冲液4 μL,5 μmol/L的正向引物0.8 μL,5 μmol/L的反向引物 0.8 μL,DNA 10 ng,2.5 mmol/L dNTP 2 μL,FastPfu聚合酶0.4 μL,加ddH2O至总体积20 μL。PCR反应条件为:95℃预变性3 min;95℃变性30 s,55℃退火30 s,72℃延伸45 s,26个循环;最终72℃延伸5 min,冷却至10℃。PCR产物用2%琼脂糖凝胶电泳检测,上样量为3 μL。
1.2.2生物信息学分析
PCR产物采用Miseq高通量测序平台进行测序。提取高质量序列进行生物信息学分析。生物信息学分析:(1)操作单元(operational taxonomic units,OTU)聚类分析:使用QIIME分析平台进行序列的生物信息学分析,与Silva数据库中已比对的16S核糖体序列数据库进行比对,对相似性在97%以上的序列进行归并,生成分类操作单元OTU;(2)菌群分类学分析:将OTU中全部序列与Silva数据库进行比对,找出其最相近且可信度达80%以上的种属信息。并将每一个OTU中的所有序列进行类比,找出同一OTU中的不同序列的最近祖先的种属信息。根据silva库中的参考序列对OTU进行种属鉴定。根据分类学比对结果,在从门至属水平上对样品中群落结构进行菌群多样性和丰度分析,采用R软件(R i386 3.0.1)进行绘图分析。
2 结果与分析
2.1微生物群落结构分析
在Phylum(门)的水平上,各组样品的微生物群落构成及其相对丰度如图1所示。
所有样品均包含Firmicutes(厚壁菌门)和Proteobacteria(变形菌门)两个主要的门,肠衣D中含有少量Bacteroidetes(拟杆菌门)。腐败香肠C含有大量的Firmicutes,占门分类上总微生物含量的94.95%,肠衣中Firmicutes的OTU占总OTU的70.11%。由图1可见,在门的水平上,各样品的微生物组成有较大差异,而正常香肠A和B的微生物组成在门的水平上差异较小。
在目(Order)的水平上,各组样品的微生物群落构成及其相对丰度如图2所示。
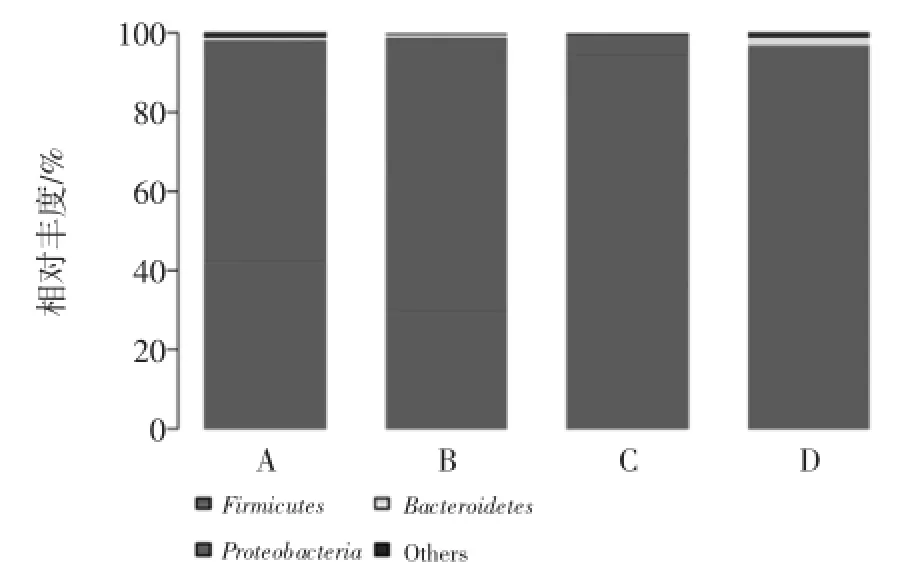
图1 香肠及肠衣样品中微生物群落在门水平上的相对丰度Fig.1 Relative abundance of bacterial phyla in microbiota of sausages and casing
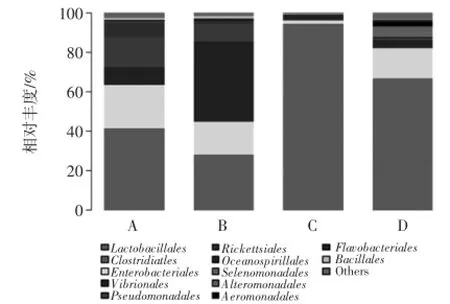
图2 香肠及肠衣样品中微生物群落在目水平上的相对丰度Fig.2 Relative abundance of bacterial order in microbiota of sausages and casing
正常香肠样品A和B中微生物多样性较高,OTU含量较高的是Lactobacillales(A:41.71%,B:28.39%)、Enterobacteriales(A:21.82%,B:16.64%)、Pseudomonadales(A:15.48%,B:8.76%)、Vibrionales(A:9.07%,B:40.80%),而腐败香肠C的微生物菌群多样性较低,丰度最高的是Lactobacillales,其OTU占总OTU的94.70%;肠衣D的微生物多样性较高,主要由Lactobacillales(25.18%)、Clostridiales(41.76%)、Enterobacteriales组成(15.51%),肠衣中还含有一定量的Vibrionales、Alteromonadales、Selenomonadales等。
在属(Genus)的水平上,各组样品的微生物群落构成及其相对丰度如图3所示。
由图3可见,在属(Genus)的水平上,A和B两种正常香肠的微生物群落中OTU含量最高的是Streptococcus、Kluyvera、Vibrio(B:40.77%)、Lactococcus、Leuconostoc等属的微生物;而腐败香肠C的微生物群落在属水平分类上与正常香肠有较大差异,腐败香肠中含量较高的微生物主要为Lactobacillus(51.19%)、Leuconostoc(36.94%),其次Weissella、Kluyvera等,根据其基因序列在种的水平上进一步与数据库已有序列比对发现,香肠C中的乳酸菌主要为Lactobacil-lus_paraplantarum,明串珠菌主要为Leuconostoc_mesenteroides_subsp._mesenteroides_J18;肠衣D中含量最高的微生物为Peptostreptococcus(37.73%),其次为Lactobacillus、Vagococcus、Plesiomonas等属的微生物。
2.2香肠及肠衣样品中微生物群落结构差异性比较
将丰度在前100位的微生物进行各组间丰度相似性聚类得到图4。
该图可通过颜色梯度及相似程度反映几种样品在属水平上群落组成的相似性及各属微生物OTU的含量。由图4可见,A和B两种香肠样品的微生物群落组成有较高的相似性,而腐败香肠C和肠衣的微生物群落与正常香肠相比有较大的差异。
基于距离矩阵进行的主坐标分析如图5所示。

图3 香肠及肠衣样品中微生物群落在属水平上的相对丰度Fig.3 Relative abundance of bacterial genus in microbiota of sausages and casing
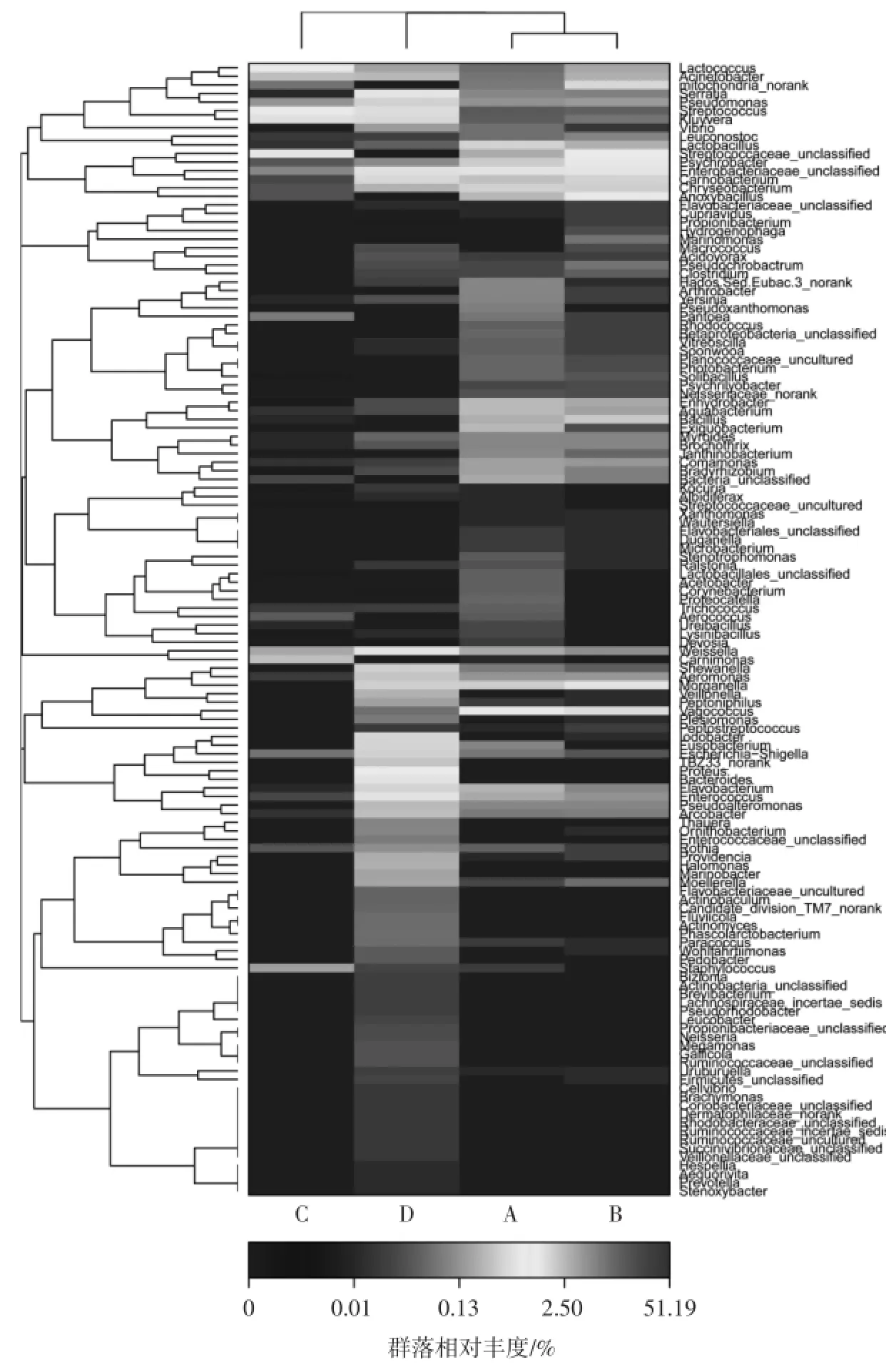
图4 香肠及肠衣样品中微生物群落热图分析Fig.4 Heatmap of sausages and casing

图5 香肠及肠衣样品微生物群落结构的主坐标分析Fig.5 PCoA plots based on unweighted Unifrac metrics
通过上述门、目及属水平的分析以及微生物群落结构的heatmap分析已初步表明不同香肠样品及肠衣的微生物群落结构有较大差异。为进一步分析其差别,对每一样品的OTU信息构建了未加权的Unifrac欧氏距离矩阵(unweighted Unifrac distance matrix)。基于这一矩阵进行了主坐标分析(Principal Co-ordinates Analysis,PCoA),由图5可见,该分析前两个主成分分别解释了总变量的49.19%和38.36%,前两个主成分共计为87.55%。在X轴的方向上,A和B香肠距离较近,表明A和B香肠的前两个主成分因素是一致的,而C和D在Y轴的方向上距离较远,说明Y因素导致C和D之间的微生物群落结构的不同。
3 讨论
本研究采用高通量测序技术分析香肠及肠衣微生物多样性,发现贮藏至不同时期的香肠及肠衣之间,微生物多样性及丰度均有较大差异。两种正常香肠微生物多样性及丰度较为相似;肠衣微生物多样性也较高,但其微生物组成与正常香肠有较大差异;而腐败香肠C微生物多样性较低,优势腐败菌由Lactobacillus(乳酸菌属)和Leuconostoc(明串珠菌属)组成。产品的微生物腐败是微生物从较低的初始菌数生长至106~107CFU/g的过程,在此期间,微生物之间通过拮抗、互惠共生或群体感应[2]等相互影响,腐败菌在增殖过程中产酸、产黏液、产生异味等导致香肠的感官方面出现腐败特性,在此过程中,具有较强竞争力并适应此种腐败环境的Lactobacillus、Leuconostoc等微生物大量生长繁殖,形成稳定的腐败菌菌相。
研究表明,真空、MPA(气调包装)及有氧条件下,肉制品中的特定腐败菌主要是乳酸菌[3-5]、肠膜明串珠菌[6-7]及 Weissella、Carnobacteriumdivergens,B.thermosphacta[8]等腐败菌。本研究与上述报道一致,但上述报道主要针对的是微生物在属水平上的分类,本研究进一步对腐败菌在种水平上进行分类,发现腐败香肠中的特定腐败菌主要为Lactobacillus_paraplantarum、Leuconostoc_mesenteroides_subsp_mesenteroides_J18,明确揭示了导致此类天然肠衣香肠腐败的微生物。
加工肉制品因原料肉的品种、成分等不同,适合生长繁殖的微生物种类也不同,腐败菌构成也因此不同。除原料肉外,辅料、香辛料及其它添加物都会带来一定种类和数量的微生物,在加工过程中不可避免地会造成二次污染,使得成品腐败微生物的菌相构成更加复杂。在贮藏过程中,腐败微生物的菌相构成发生改变,其中一种或几种微生物会成为优势菌群,最终成为导致肉和肉制品腐败的优势腐败菌;而其它种类则数量较少,甚至在相互竞争中逐渐消亡。这就是本研究中几种类型香肠及肠衣微生物多样性发生重大变化的原因,到贮藏后期,真正起主导作用的只是Lactobacillus_paraplantarum、Leuconostoc_mesenteroides_ subsp_mesenteroides_J18,其它微生物在竞争中消亡。但此两种微生物在初始微生物中含量较低(OTU所占比例均在1.00%以下),是何原因导致这两种微生物在腐败香肠中占据了绝对的优势,仍需结合微生物代谢学及环境微生物学等做进一步的研究。其腐败规律的揭示,将对在生产及贮藏中减少天然肠衣香肠的微生物污染,延长产品的货架期有着重要的意义。
参考文献:
[1]Miambi E,Guyot JP,Ampe F.Identification,isolation and quantification of representative bacteria from fermented cassava dough using an integrated approach of culture-dependent and culture-independent methods[J].International Journal of Food Microbiology,2003,82(2):111-120
[2]Ammor MS,Michaelidis C,Nychas G-JE.Insights into the role of quorum-sensingin food spoilage[J].Journal of Food Protection,2008,71(7):1510-1525
[3]Borch E,Kant-Muermans ML,Blixt Y.Bacterial spoilage of meat and cured meat products[J].International Journal of Food Microbiology,1996,33(1):103-115
[4]Samelis J,Kakouri A,Rementzis J.Selective effect of the product type and the packaging conditions on the species of lactic acid bacteria dominating the spoilage microbial association of cooked meats at 4℃[J].Food Microbiology,2000,17(3):329-340
[5]Pothakos V,Devlieghere F,Villani F,et al.Lactic acid bacteria and their controversial role in fresh meat spoilage[J].Meat Science,2015,109:66-74
[6] Hastings JW,Stiles ME,vonHoly A.Bacteriocins of Leuconostocs isolated from meat[J].International Journal of Food Microbiology,1994,24(1/2):75-81
[7]Hu P,Zhou GH,Xu XL,et al.Characterization of the predominant spoilage bacteria in sliced vacuum-packed cooked ham based on 16S rDNA-DGGE[J].Food Control,2009,20(2):99-104
[8]Korkeala HJ,Björkroth KJ.Microbiological spoilage and contamination of vacuum-packaged cooked sausages[J].Journal of Food Protection,1997,51(6):302-303
DOI:10.3969/j.issn.1005-6521.2016.11.027
基金项目:国家自然科学基金(31501442);天津市重点支撑项目(14ZCZDNC00015);公益性行业(农业)科研专项经费项目(201303082-3);天津科技大学青年教师创新基金(2014CXLG03)
作者简介:滕安国(1982—),男(汉),实验师,硕士,研究方向:肉品加工与质量控制。
*通信作者:张芹(1981—),女(汉),助理研究员,硕士,研究方向:食品微生物。
收稿日期:2016-03-28
Application of High-throughput Sequencing to Analyze Microbial Composition of Sausages with Natural Casing
TENG An-guo,QI Xiao-na,ZHANG Qin*,WANG Wen-hang
(College of Food Engineering and Biotechnology,Tianjin University of Science and Technology,Tianjin 300457,China)
Abstract:Microbial diversity and abundance among sausages with natural casing stored to different periods were analyzed by Miseq high-throughput sequencing technology.The results showed that the microbial diversity and abundance were relatively high in normal sausages and natural casing.However,the microbial diversity in spoilage sausage was low,the bacteria enriched in spoilage sausage were from genus Lactobacillus and Leuconostoc.Classified at the level of species,Lactobacillus_paraplantarum and Leuconostoc_mesenteroides_subsp_mesenteroides_J18 were the dominant bacteria in spoilage sausage.
Key words:sausages with natural casing;microbial diversity;high-throughput sequencing
