变异链球菌ldh基因启动子的克隆及活性检测
2016-07-27张加勤黄珊珊徐巧丽饶慧华马晓波黄朝阳房丽丽郑港森宋秀宇
张加勤,黄珊珊,2,徐巧丽,饶慧华,马晓波,黄朝阳,房丽丽,郑港森,宋秀宇
变异链球菌ldh基因启动子的克隆及活性检测
张加勤1,黄珊珊1,2,徐巧丽1,饶慧华1,马晓波1,黄朝阳1,房丽丽1,郑港森1,宋秀宇3
1.厦门大学附属第一医院暨福建医科大学厦门市第一教学医院检验科,厦门361003;2.福建医科大学第一临床医学院,福州350108;3.厦门市中心血站,厦门361003
摘要:目的克隆变异链球菌ldh基因启动子,并验证其活性。方法以变异链球菌UA159基因组为模版,PCR扩增变异链球菌ldh基因候选启动子,将其插入β-葡萄糖醛酸酶 (β-glucuronidase,gusA)报告基因表达载体pIB107 BamH I/Xho I之间,构建ldh基因启动子gusA报告载体pCKS11,PCR、酶切及测序鉴定;经Sca I酶线性化后转化变异链球菌UA159,卡那霉素筛选阳性克隆SCKS11,经PCR和测序鉴定后,检测其GusA活性。结果成功扩增出大小为269 bp的ldh基因候选启动子;经PCR、酶切及测序鉴定,变异链球菌ldh基因候选启动子gusA报告基因表达载体pCKS11构建正确;PCR和测序鉴定,ldh基因候选启动子gusA报告株SCKS11构建成功;变异链球菌ldh基因候选启动子启动的GusA活性是无启动子的阴性对照的5.8倍,是阳性对照变异链球菌clpP基因启动子的0.9倍。结论成功克隆变异链球菌ldh基因启动子序列,具有较强的启动转录活性,为研究ldh基因的表达调控机制奠定基础。
关键词:变异链球菌;ldh基因;启动子
Supported by the National Natural Science Foundation of China (No. 81000762) and the Natural Science Foundation of Fujian Province (Nos. 2015J0155 and 2013D002)
变异链球菌(Streptococcusmutans,S.mutans)是人类口腔的主要致龋菌之一。 变异链球菌在牙面粘附定植形成复杂的被称为牙菌斑的生物膜结构,并在牙菌斑内代谢碳水化合物产酸,造成局部微环境PH值下降,牙齿硬组织脱矿,羟磷灰石溶解破坏致龋[1]。其致龋的毒力作用主要表现在粘附、产酸和耐酸三个方面。除此之外还可引起牙髓病、牙周病、颌骨炎、感染性心内膜炎等一系列并发症,严重影响人类的身心健康。
变异链球菌属兼性厌氧革兰氏阳性球菌,其能量来源于无氧糖酵解途径。乳酸脱氢酶(lactate dehydrogenases,LDH)是无氧糖酵解途径中重要的酶,广泛存在于各种生物体中,也是S.mutans能量代谢途径中不可缺少的固有酶,在无氧糖酵解途径中,可催化乳酸和丙酮酸相互转换,调节生物体内酸代谢的平衡。为研究S.mutansldh基因启动子的结构和ldh基因的表达调控机制,我们构建了S.mutansldh基因5′侧翼序列-β-葡萄糖醛酸酶 (β-glucuronidase,gusA)报告基因表达载体,通过同源重组构建了ldh-gusA基因表达报告株,并测定其活性,进而判断启动子活性,为研究ldh基因的表达调控机制奠定基础。
1材料与方法
1.1菌株和质粒变异链球菌UA159由第四军医大学馈赠;质粒pIB107由堪萨斯大学医学中心 Biswas Indranil 教授惠赠;clpP基因启动子gusA报告基因表达载体pFclpP及其报告株SFclpP由本实验室构建并保存;大肠埃希菌DH5α和大肠埃希菌Top10为本实验室保存。
1.2主要试剂T4 DNA连接酶、限制性内切酶、DL2000 DNA Marker 、DL5000 DNA Marker、1 kb DNA Ladder购自TaKaRa公司;感受态刺激肽CSP、 SanPrep柱式质粒DNA小量抽提试剂盒购自上海生工生物工程技术服务有限公司;2×Taq PCR master Mix试剂盒、细菌基因组DNA提取试剂盒购自北京天根生化科技公司;PCR产物/凝胶回收纯化试剂(QIAquick PCR Purification Kit及QIAquick Gel Purification Kit)购自QIAGEN公司;p-nitrophenyl-β-D-glucoside (PNPG)购自Sigma公司;其余为国产或进口分析纯试剂;所有引物合成和DNA测序均由北京六合华大基因科技股份有限公司完成。
1.3实验步骤
1.3.1细菌基因组DNA的提取取37 ℃静置过夜培养变异链球菌,按照细菌基因组DNA提取试剂盒说明书提取变异链球菌UA159基因组DNA。
1.3.2目的片段的扩增以变异链球菌UA159基因组DNA为模版,设计引物BamH I-ldh-p-F1(5′- CGGGATCCCCGAGCAACAATAACACTC-3′)和XhoI-ldh-p-R1(5′-CCGCTCGAGAACATCTCCTTATAATTTATTAAGTATATATT
CTAT-3′)扩增ldh基因候选启动子。在200 μL PCR反应管中建立如下反应体系:UA159基因组DNA 50 ng,2×Taq PCR Master Mix 12.5 μL,上、下游引物各0.5 μL(10 μmol/L),加灭菌水至25 μL。反应条件:预热95 ℃预变性2 min;95 ℃变性30 s,57 ℃退火30 s,72 ℃延伸30 s,共30循环;72 ℃延伸10 min。PCR产物经1%琼脂凝胶电泳120 V,30 min后观察结果,并进行PCR产物纯化回收。
1.3.3ldh基因候选启动子重组gusA报告载体的构建与鉴定 变异链球菌ldh基因候选启动子序列经QIAquick PCR Purification Kit回收纯化后,限制性内切酶BamH I/XhoI酶切,插入经BamH I/XhoI酶切的gusA报告基因表达载体pIB107,转化大肠埃希菌DH5α感受态细胞。用含氨苄西林(100 μg/mL)和卡那霉素(50 μg/mL)的固体培养基筛选阳性菌落,37 ℃过夜培养并抽提质粒获得同源重组报告载体pCKS11,用引物BamH I-ldh-p-F1/XhoI-ldh-p-R1进行PCR验证,并同时进行酶切及测序鉴定。
1.3.4变异链球菌ldh基因候选启动子重组gusA报告基因表达株构建 将含有ldh基因候选启动子的同源重组gusA报告基因表达载体pCKS11,经限制性内切酶ScaI线性化后,参考文献[2]转化变异链球菌UA159,卡那霉素(300 μg/mL)筛选阳性菌落,构建ldh启动子片段gusA报告基因表达株SCKS11。提取细菌基因组,设计引物kan-F2(5′-GGCAATCTGCCTCCTCATC-3′)/gusA-In-R2(5′-CTGCCTGGCACAGCAATTGCCCGGC-3′),经PCR及测序验证。
1.3.5变异链球菌ldh基因候选启动子序列活性检测参照文献[3]分别测定ldh基因候选启动子gusA报告基因表达株SCKS11、clpP基因启动子gusA报告基因阳性对照株SFclpP和无启动子的阴性对照株SIB107的GusA蛋白活性,鉴定变异链球菌ldh基因候选启动子活性。37 ℃增菌培养至对数生长期,记录OD600值。将离心后的沉淀物用磁珠法破碎细胞提取上清蛋白液。酶标板中分别加Z-buffer 50 μL,各自细胞裂解液50 μL和PNPG溶液50 μL,混匀,每个样本做2孔。待活性达到稳定,溶液变成亮黄色后,加入1 mol/L Na2CO350 μL,用分光光度计测定A405值,每个样本测3次。用公式[1 000×A405]/[time(min) ×cell OD600]计算GusA活性,单位为MU。
2结果
2.1变异链球菌ldh基因候选启动子的体外扩增 采用引物BamH I-ldh-p-F1/XhoI-ldh-p-R1 PCR扩增变异链球菌ldh基因候选启动子序列,产物经1 %琼脂糖凝胶电泳分析,在281 bp处有特异性扩增条带,大小与预期结果一致,且未见非特异扩增带。(如图1)。
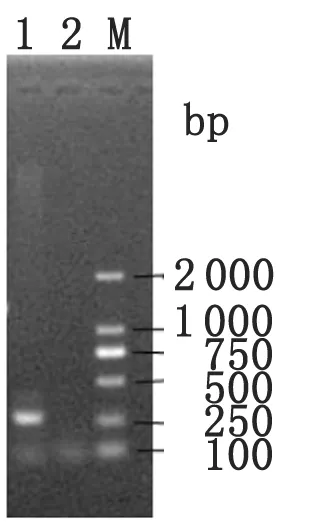
1:变异链球菌ldh基因候选启动子序列PCR扩增片段;2:阴性对照;M: DNA Marker DL2 000
1: PCR product of candidate promoter ofS.mutansldhgene; 2: Negative control; M: DNA Marker DL2 000
图1变异链球菌ldh基因候选启动子PCR扩增结果
Fig.1PCR analysis of candidate promoter of S. mutans ldh gene
2.2变异链球菌ldh基因候选启动子gusA报告基因表达载体的构建与鉴定变异链球菌ldh基因候选启动子经BamH I/XhoI酶切,插入到经BamH I/XhoI酶切的pIB107载体中,获得ldh基因候选启动子gusA报告基因表达载体pCKS11。转入DH5α感受态细胞后,提取质粒,用BamH I-ldh-p-F1/XhoI-ldh-p-R1引物进行PCR扩增,PCR产物经1%琼脂糖凝胶电泳分析显示在281 bp处有特异性扩增条带;质粒pCKS11经ClaI单酶切和BamH I/PstI双酶切,酶切产物经1%琼脂糖凝胶电泳分析显示:ClaI单酶切片段在2 246 bp和6 689 bp位置出现两条带,BamH I /PstI双酶切片段大小为2 191 bp和6 744 bp,大小与预期结果一致。测序结果证明成功将ldh基因候选启动子插入pIB107gusA报告基因表达载体,序列与预期结果一致。(如图2)。
2.3变异链球菌ldh基因候选启动子gusA报告基因表达株的构建与鉴定 变异链球菌ldh基因候选启动子gusA报告基因表达载体pCKS11经线性化后转入变异链球菌UA159,在卡那霉素(300 μg/mL)抗性的THY平板上长出阳性菌株,说明变异链球菌ldh基因候选启动子gusA报告基因以及卡那霉素抗性基因成功整合到变异链球菌UA159的基因组中。提取ldh基因候选启动子gusA报告基因表达株基因组DNA,用引物Kan-F2/GusA-In-R2建立PCR扩增体系,电泳后,结果显示分别在1 091 bp和822 bp处有特异性扩增条带,与预期结果大小相符(如图3)。测序结果证明变异链球菌kan抗性基因盒-ldh基因候选启动子-gusA报告基因已经插入变异链球菌转座酶基因(SMU.1405)中,成功构建ldh候选启动子gusA报告基因表达株SCKS11。
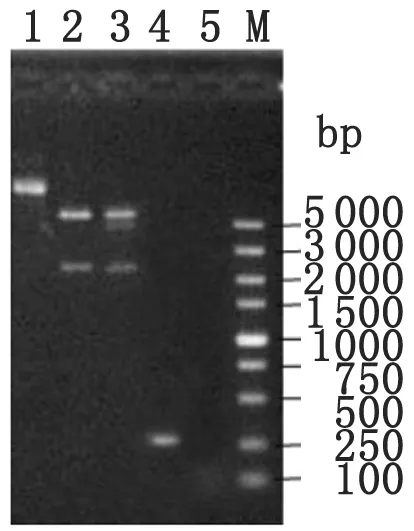
1: pCKS11质粒; 2: pCKS11质粒ClaI单酶切鉴定;3: pCKS11质粒BamH I /PstI双酶切鉴定;4: pCKS11质粒PCR鉴定;5: 阴性对照; M: DNA Marker DL5 000
1:Recombinant plasmid pCKS11; 2:ClaI digestion of recombinant plasmid pCKS11; 3:BamH I /PstI digestion of recombinant plasmid pCKS11; 4: PCR product of recombinant plasmid pCKS11; 5: Negative control; M: DNA Marker DL5 000
图2变异链球菌ldh基因候选启动子gusA报告基因表达载体的鉴定
Fig.2Verification of gusA report vector of ldh gene candidate promoter of S. mutans

1:线性化pIB107质粒gusA报告基因表达株的PCR扩增产物; 2:阴性对照;3:线性化pCKS11质粒gusA报告基因表达株的PCR扩增产物; 4:阴性对照;
M: DNA Marker DL2 000;1: PCR product of strains with linearized gusAreport gene plasmid pIB107; 2: Negative control; 3: PCR product of strains with linearized gusAreport gene plasmid pCKS11; 4: Negative control; M: DNA Marker DL2 000
图3变异链球菌ldh基因候选启动子gusA报告基因表达株的PCR鉴定
Fig.3Validation of the gusA report strains of ldh gene candidate promoter of S. mutans by PCR
2.4变异链球菌ldh基因候选启动子GusA活性测定结果显示ldh基因候选启动子gusA报告基因表达株SCKS11的GusA活性是无启动子的阴性对照株SIB107的5.8倍,是clpP基因启动子gusA报告基因阳性对照株SFclpP的0.9倍,表明成功克隆ldh基因启动子,且该启动子有较强启动基因转录活性。详见图4。
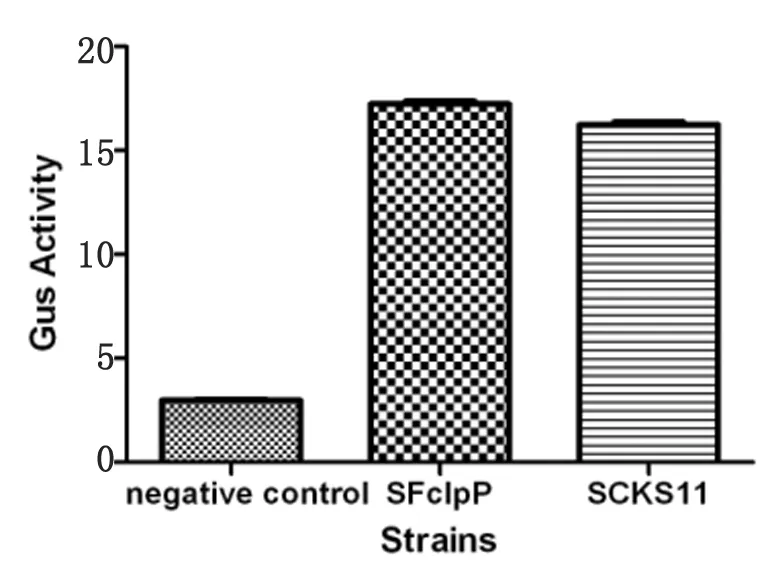
Negative control SIB107 (pIB107); SFclpP (pFclpP); SCKS11 (pCKS11)
图4变异链球菌ldh基因候选启动子GusA活性检测
Fig.4GusA activity analysis of ldh gene candidate promoter of S. mutans
3讨论
无氧糖酵解途径中,乳酸脱氢酶催化丙酮酸形成乳酸,是变异链球菌糖代谢过程中极为重要的酶,为细菌的生长代谢提供能量,并且该酶在细菌体内稳定表达[4]。最早,由ldh基因编码的乳酸脱氢酶LDH是由M.J.Duncan等在变异链球菌中分离纯化出的细菌体内固有酶,菌内含量高。正由于LDH在菌体无氧糖酵解过程中的重要作用,其可作为代谢活性的指示剂[5]。同时,在原核生物中,由于细菌RNA聚合酶不能识别真核基因的启动子,因此采用变异链球菌ldh基因的启动子构建报告质粒,使其可在原核生物中转录翻译,从而监测ldh基因的表达情况。
转录翻译是原核生物基因的主要调控方式,通过RNA聚合酶、转录因子、启动子和终止子的相互作用实现转录调控,改变细胞表型,从而实现细胞生理状态的改变。原核生物的启动子是结构基因上游的一段DNA序列,本研究所选取的ldh基因候选启动子序列在转录起始点到其上游269 bp,而一般是从翻译起始位点到其上游200 bp左右,说明所选取的序列为启动子的可能性较高。启动子的核心元件主要包括RNA聚合酶的识别和结合位点、间隔区、SD序列和转录起始位点,在转录翻译中起重要作用。据生物信息学软件分析结果显示,在S.mutans中,ldh基因翻译起始点上游-79 bp~-74 bp 处有在RNA聚合酶在σ亚基的协助下被RNA聚合酶识别结合的序列5′-GTGACT-3′,形成封闭的RNA聚合酶全酶-启动子复合物。与通常被认为最优化-35区序列5′-TTGACA-3′相比,同源性67 %,认为其是-35区的可疑序列;分析显示,在S.mutans中,ldh基因翻译起始点上游的-20 bp~-15 bp处有与RNA聚合酶的核心酶紧密结合形成开放性启动子复合物的序列 5′-TATAAG-3′。形成的开放性启动子复合物可将DNA解链,当RNA聚合酶移动到转录起始点时,加入三磷酸核苷酸开始转录[6-7]。此序列与-10区最优化序列5′-TATAAT-3′相比同源性83 %,研究发现第1,2,6位三个碱基最保守,但是对启动子的识别和解链作用主要在第2位的A碱基上,说明此序列为-10区的可能性较高。通过分析结果,我们发现翻译起始位点上游-17 bp~-13 bp处,有强SD序列的偏好模式5′-AAGGA-3′,可与核糖体16s rRNA 3′末端富含嘧啶序列互补。研究表明,SD序列越强,基因表达水平越高。综上所述,ldh基因候选启动子有启动子的特征可作为候选启动子。成功定位启动子的区域是研究启动子,构建报告基因表达载体的关键步骤。
生物工程领域通常选用由报告基因、抗性基因和转录终止子构成,但缺乏启动子的克隆表达载体为报告基因载体,从而研究某个基因在菌体内的表达情况和对其他基因的相互作用影响[8]。目前,基因工程领域常用的报告基因有cat(氯霉素乙酰转移酶基因)、lacZ(β-半乳糖苷酶基因)、lux(荧光素酶基因)、gusA(β-葡萄糖醛酸酶基因)和荧光蛋白等。cat基因和lux基因的表达产物分析成本高,lacZ基因的表达背景高,荧光蛋白不能定量测定,而gusA基因编码的β-葡萄糖醛酸酶能水解多种β-葡萄糖醛酸衍生物,且GusA活性可以用分光光度法、荧光光度法、组织化学法等不同方法检测分析。本实验采用分光光度法检测gusA基因编码的β-葡萄糖醛酸酶,具有较高灵敏度、不需要昂贵的仪器设备和操作简便等优点。
为了研究变异链球菌ldh基因启动子的活性,本研究克隆ldh基因候选启动子,采用Indranil Biswas 课题组所构建的pIB107gusA报告基因表达载体,成功构建了变异链球菌ldh-gusA报告株,并证实了ldh基因启动子的活性。该报告株的优点是将kan抗性基因盒-ldh基因候选启动子-gusA报告基因整合到变异链球菌的染色体DNA中,既可稳定表达目的基因,又可通过kan抗性基因筛选阳性菌株进行GusA活性检测。pIB107gusA报告基因表达载体与变异链球菌发生同源重组原理是pIB107载体的卡那霉素抗性基因盒的5′侧翼和gusA报告基因及3′侧翼序列分别为变异链球菌转座酶基因(SMU1405)5′端及3′端同源臂[3]。线性化的载体可以通过同源重组的方法将同源臂整合到变异链球菌转座酶基因上,那么,卡那霉素基因和gusA报告基因就被重组到变异链球菌基因组上,用卡那霉素抗性基因可筛选出阳性菌落。同法,将ldh基因启动子克隆到pIB107的gusA报告基因上游,卡那霉素基因和gusA报告基因连同ldh基因启动子就可被重组到变异链球菌基因组上,整合到基因组上的ldh基因启动子启动gusA报告基因,转录翻译GusA蛋白即β-葡萄糖醛酸酶。β-葡萄糖醛酸酶催化水解底物PNPG,产生黄色水解产物对-硝基苯酚,终止反应后,可用分光光度计或酶标仪检测酶反应所释放的对-硝基苯酚的A405值,用公式计算GusA活性,通过此方法测定GusA活性从而判断插入的启动子活性[9]。从研究结果中可看出,ldh基因候选启动子gusA报告基因表达株SCKS11的启动子活性是无启动子的阴性对照的5.8倍,是阳性对照变异链球菌clpP基因启动子的0.9倍,说明ldh基因启动子具有较强的启动基因转录活性。
变异链球菌ldh基因启动子的成功克隆和ldh-gusA报告株的成功构建为后续研究ldh基因启动子的结构和功能提供了生物模型和实验依据,并且将为研究ldh基因的表达调控机制奠定基础。
参考文献:
[1]Singh K, Senadheera DB, Levesque CM, et al. The copYAZ operon functions in copper efflux, biofilm formation, genetic transformation, and stress tolerance inStreptococcusmutans[J]. J Bacteriol, 2015, 197(15): 2545-2557.
[2]Ahn SJ, Kaspar J, Kim JN, et al. Discovery of novel peptides regulating competence development inStreptococcusmutans[J]. J Bacteriol, 2014, 196(21): 3735-3745.
[3]Biswas I, Jha JK, Fromm N. Shuttle expression plasmids for genetic studies inStreptococcusmutans[J]. Microbiology, 2008, 154(Pt 8): 2275-2282.
[4]Baker JL, Derr AM, Faustoferri RC, et al. Loss of NADH oxidase activity inStreptococcusmutansleads to rex-mediated overcompensation in NAD+ regeneration by lactate dehydrogenase[J]. J Bacteriol, 2015, 197(23): 3645-3657.
[5]Merritt J, Kreth J, Qi F, et al. Non-disruptive, real-time analyses of the metabolic status and viability ofStreptococcusmutanscells in response to antimicrobial treatments[J]. J Microbiological Methods, 2005, 61(2): 161-170.
[6]Osterberg S, Peso-Santos TD, Shingler V. Regulation of alternative sigma factor use[J]. Annual Rev Microbiol, 2011, 65(1): 37-55.
[7]Islam MS, Pallen Mark J, Busby Stephen JW. A cryptic promoter in the LEE1 regulatory region of enterohaemorrhagicEscherichiacoli: promoter specificity in AT-rich gene regulatory regions[J]. Biochem J, 2011, 436(3): 681-686.
[8]Bron PA, Hoffer SM, Van Swam II, et al. Selection and characterization of conditionally active promoters in Lactobacillus plantarum, using alanine racemase as a promoter probe[J]. Appl Environmental Microbiol, 2004, 70(1): 310-317.
[9]Zhang J, Banerjee A, Biswas I. Transcription of clpP is enhanced by a unique tandem repeat sequence inStreptococcusmutans[J]. J Bacteriol, 2009, 191(3): 1056-1065.
DOI:10.3969/j.issn.1002-2694.2016.03.003
通讯作者:宋秀宇,Email:songxyxm@126.com
中图分类号:R378.1
文献标识码:A
文章编号:1002-2694(2016)03-0224-05
Corresponding author:Song Xiu-yu, Email: songxyxm@126.com
收稿日期:2015-11-26;修回日期:2016-01-06
Activity detection of ldh gene promoter of Streptococcus mutans
ZHANG Jia-qin1,HUANG Shan-shan1,2,XU Qiao-li1,RAO Hui-hua1,MA Xiao-bo1,HUANG Chao-yang1,FANG Li-li1,ZHENG Gang-sen1,SONG Xiu-yu3
(1.DepartmentofClinicalLaboratory,theFirstAffiliatedHospitalofXiamenUniversity,Xiamen361003,China;2.TheFirstClinicalMedicalCollegeofFujianMedicalUniversity,Fuzhou350108,China;3.XiamenCentralBloodServiceStation,Xiamen361003,China)
Abstract:We cloned the ldh gene promoter of Streptococcus mutans and explored it′s activity. The candidate promoter of S. mutans ldh gene was amplified from S. mutans UA159 chromosome DNA by PCR, then was inserted into pIB107 by BamH I/Xho I to construct β-glucuronidase report plasmids pCKS11. The plasmids pCKS11 were verified by PCR, restriction enzyme and sequencing. After being linearized by Sca I, the plasmids pCKS11 were transformed into S. mutans UA159. Then, the strain was screened out by THY agar contain 300 μg/mL kanamycin. After being confirmed by PCR and sequencing, the GusA activity of homologous recombination β-glucuronidase report strain SCKS11 and it parental strains were measured. Results showed that the 269 bp candidate promoter of ldh gene was amplified successfully. PCR, restrictive endonuclease digest and sequencing confirmed the validity of the ldh-gusA report plasmids and strains. The result of GusA assays showed that the activity of the candidate promoter of S. mutans ldh gene were 5.8 folds of that of the negative control without any promoter and 0.9 folds of that of the clpP gene promoter of S. mutans positive control. The success of locating the promoter of S. mutans ldh gene and construction of report strains offered an experimental basis to the further study of the expression and regulation of ldh gene of S. mutans.
Keywords:Streptococcusmutans ; ldh; promoter
国家自然科学基金(No.81000762)与福建省自然科学基金(No.2015J0155,2013D002)联合资助
