头孢呋辛酸的合成研究
2016-07-26曹卫凯
曹卫凯
(西安万隆制药股份有限公司,西安 710119)
头孢呋辛酸的合成研究
曹卫凯
(西安万隆制药股份有限公司,西安 710119)
摘 要从7-氨基头孢烷酸(7-ACA)出发合成头孢呋辛酸,同时对合成过程及关键点进行优化。首先7-ACA在甲醇和水的混合溶剂中低温水解,生成的D-7-ACA,不经分离,直接进行7-位酰化反应,经结晶分离干燥后得到关键中间体DCC(去氨甲酰基头孢呋辛酸),收率可达84.8%。后者经过与氯磺酸异氰酸酯(CSI)反应完成C-3位的氨甲酰化改造得到头孢呋辛酸的溶液,结晶分离干燥后,收率可达91.4 %,合计收率达77.5 %。该工艺具有收率高、低成本、易操作等特点,极具有工业化生产价值。
关键词头孢菌素;头孢呋辛;合成;工业化
头孢呋辛是由英国葛兰素公司研发的一个二代头孢类抗生素,1988年美国上市,由于其具有广谱抗菌作用、肾脏毒性低,对多种革兰氏阳性和革兰氏阴性细菌有效,同时对β-内酰胺酶稳定等特点[1],被广泛应用于对抗敏感菌引起的各类感染。其抗菌作用机制是该药物通过与细菌细胞膜上的青霉素结合蛋白(PBPs)结合,抑制细胞分裂和生长,最后使细菌溶解和死亡[2-3]。市面上目前使用制剂为头孢呋辛钠(粉针剂)和头孢呋辛酯(片剂),头孢呋辛为两种制剂的活性成分(API),前者注射用,后者用于口服,主要用于治疗敏感菌所致的呼吸道感染、耳、鼻、喉科感染、泌尿道感染、皮肤和软组织感染、骨和关节感染、淋病、包括败血症及脑膜等其他感染。该产品己成为抗感染药物中的一线用药,目前为国家基本药物目录品种。
文献报道的头孢呋辛合成路线有多条,主要的工艺路线有以下几种:
(1)以7-ACA为起始物,溶解后,进行7位氨基酰化,之后3位水解,得到3-去氨甲酰基-头孢呋辛酸,再将3位羟甲基改造得到头孢呋辛酸[4-6]。
(2)以7-氨基头孢烷酸(7-ACA)为起始物,水解得(3-去乙酰基-7-氨基-头孢烷酸)D-7-ACA,然后将3位的羟甲基改造为氨甲酰氧甲基,再将7位氨基酰化引入侧链甲氧亚胺基呋喃乙酸铵盐(SMIA)得到头孢呋辛酸[7]。
(3)以7-ACA为起始物,水解得D-7-ACA,先进行7位氨基的酰化将侧链引入,得到3-去氨甲酰基-头孢呋辛酸(DCC),再进行3位的羟甲基改造,同样可得头孢呋辛酸[8-13]。
其中,方法(1)采用先7位氨基酰化,得到酰胺,再在碱性条件下进行3位酯的水解,但3位酯水解的同时,7位的酰胺键也会在碱性条件下发生部分水解,这样会使得到的中间体3-去氨甲酰基-头孢呋辛酸里面含有大量水解杂质,水解杂质包括D-7-ACA、SMIA,最终使得3-去氨甲酰基-头孢呋辛酸的纯度大大降低;方法(2)采用3位氨甲酰氧甲基改造后,再进行7位酰化,虽然避免方法(1)中酰胺被水解的不足,但由于酰化时都是将SMIA先与三氯氧磷反应,制成有活性的酰氯,再与7位氨基反应,由于3位已经经过改造,含有裸露的氨基,该方法避免不了酰氯与3位氨甲酰氧甲基上的氨基的反应,使成品中含有大量3位氨基被酰化的杂质,导致最终头孢呋辛酸纯度降低。方法(3)是经过先水解3位酯,再酰化7位,最后改造3位,这样不但可以避免方法(1)中酰胺被水解的不足,同时可避免方法(2)中3位氨基的酰化。
综上,方法(3)能有效地规避方法(1)和(2)的不足,本研究以方法(3)为基础,7-ACA以水解得到D-7-ACA,产物不经分离,直接在水相中与7位侧链酰氯进行混合相反应,制得3-去氨甲酰基-头孢呋辛酸(DCC),DCC经分离、干燥后与CSI反应,水解得到头孢呋辛酸溶液,之后进行结晶、过滤、干燥后可得头孢呋辛酸成品。
1 实验部分
1.1 主要仪器与试剂
JD 1000-2型电子称;德国IKA-RW20型电动搅拌机;梅特勒FE20K Plus型酸度计;DLSB-30/-40型低温循环泵;ZDJ-3S型自动水分测定仪;岛津LC-2010 CHT型高效液相色谱仪。7-ACA、SMIA、CSI均为工业级,其余试剂为分析纯。
1.2 实验方法
1.2.1 甲氧亚胺基呋喃乙酰氯(Ⅳ)的合成
在250 mL三口反应瓶中加入DMF(30 g,0.41 mol)、DMAC(36 g,0.41 mol)和Ⅲ SMIA(17 g,0.091 mol),之后再加入DMC(40 mL),在搅拌下降温到-20 ℃,加入三氯氧磷(16.9 g,0.110 mol),控温-15~ -10 ℃,反应120 min,得含Ⅳ反应液,降温至-30±5 ℃备用。合成过程见图1。
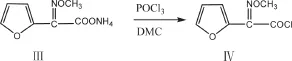
图 1 Ⅳ的合成路线Fig.1 Synthetic route of Ⅳ
1.2.2 3-去氨甲酰基-头孢呋辛酸DCC(Ⅴ)的合成
在500 mL三口反应瓶中加入甲醇(120 mL)、水(80 mL)和Ⅰ7-ACA(24 g,0.088 mol),搅拌下降温至-35 ℃,滴加10 %氢氧化钠(60 mL),搅拌30 min后,在25 min内滴加10 %盐酸(20 mL),加入备用酰氯Ⅳ,反应30 min后,加入DMC(90 mL)进行洗涤,向水相中滴加10 %盐酸,调pH到2.0,过滤,滤出固体用水洗涤,真空干燥得淡黄色粉末DCC(28.5 g,0.074 mol,收率84.8 %)合成过程见图2。
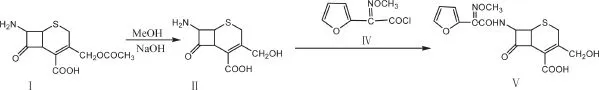
图 2 Ⅴ的合成路线Fig.2 Synthetic route of Ⅴ
1.2.3 头孢呋辛酸(Ⅵ)的合成
在500 mL三口瓶反应中加入乙腈(97 mL),启动搅拌降温到-30 ℃,加入中间体DCC(28.5 g,0.074 mol),控温至-30~-20 ℃,加入CSI(15 g,0.106 mol),保温-26~-24 ℃,反应80 min,加入水(40 mL),反应180 min,再向反应液中加入(200 mL)水,控温15~25 ℃,滴加氨水,在45 min内,调节pH到5.6,加入DMC(20 mL)萃取分层,向水层中加入10 %盐酸,控制搅拌转速,调pH到 2.0,过滤,滤出固体用水洗涤,真空干燥得头孢呋辛酸(29 g,0.068 mol,收率91.4 %)。头孢呋辛酸合成过程见图3。
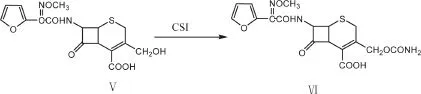
图 3 头孢呋辛酸的合成路线Fig.3 Synthetic route of cefuroxime
2 结果与讨论
2.1 酰氯化试剂三氯氧磷的用量对收率的影响
由表1可知:呋喃铵盐与三氯氧磷配比为1∶1.2时,最为适宜,当三氯氧磷用量少时反应不完全,收率低;随着三氯氧磷的增多,收率变化很小,同时由于三氯氧磷投入量太大会造成过量的三氯氧磷对环境造成严重危害及生产三废难处理等问题,故三氯氧磷量优选呋喃铵盐的1.2倍。
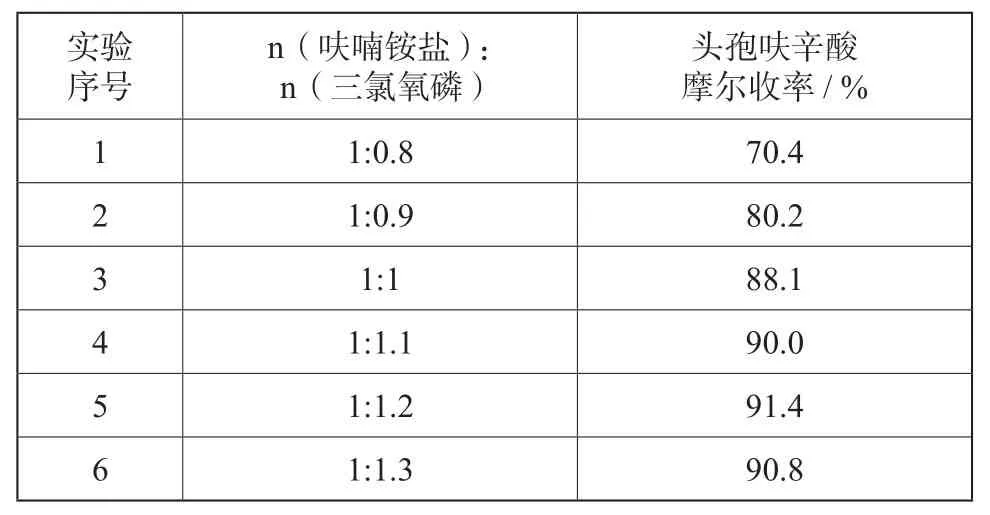
表 1 三氯氧磷用量对反应收率的影响Tab. 1 Effect of dosage of POCl3 on the yield of product
2.2 DCC制备过程中盐酸加料时间对收率的影响
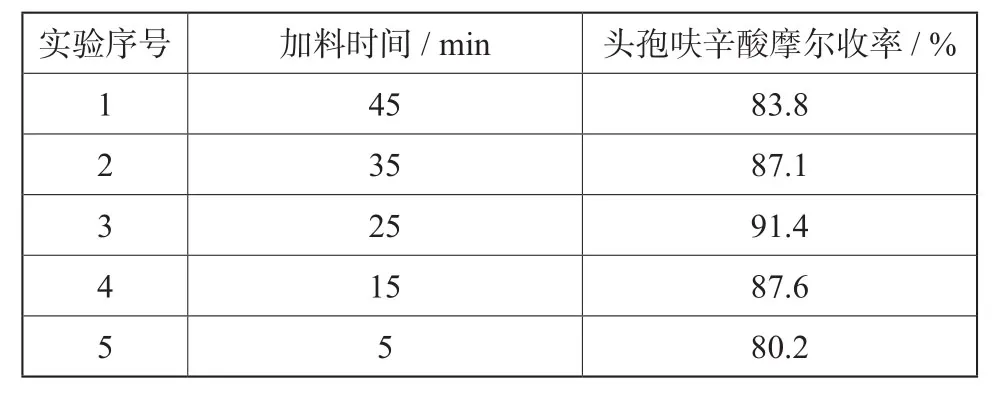
表 2 盐酸加料时间对反应收率的影响Tab. 2 Effect of dropping time on the yield of product
由表2可知:加料时间为25 min时,最为适宜。对于盐酸加料时间缩短时,呋辛酸的收率变高,但当再要缩短时间时,就会适得其反,头孢呋辛酸的收率会大大降低,这是由于在酸性条件下D-7-ACA的羧基可与其3位的羟基缩合形成内酯(杂质产生反应式见下图4),导致中间体DCC纯度降低,而使最终成品的收率降低。
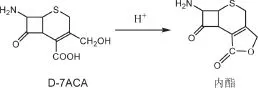
图 4 杂质形成机理Fig.4 Mechanism for impurity forming
2.3 呋辛酸缩合时反应温度对收率影响
在其他条件不变的情况下,只改变缩合反应温度,观察对反应收率的影响,具体实验数据见表3。
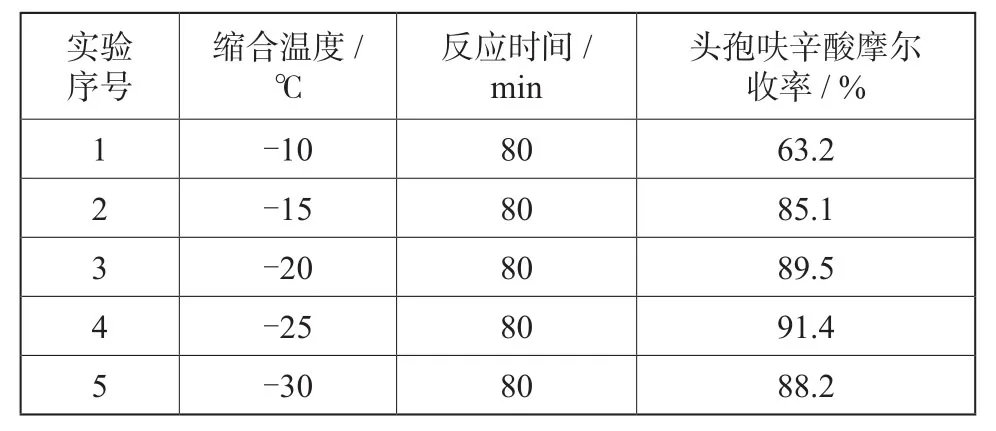
表3 呋辛酸缩合反应温度对收率的影响Tab. 3 Effect of different reaction temperature on the yield of product
由表3可知,实验4较为理想,在此情况下,头孢呋辛酸的收率较高,实验1,由于反应温度太高,反应产生杂质太多,最终导致产品收率大幅下降;对于实验5,由于反应温度太低,在固定的时间内反应率较低,但在该温度下,笔者通过延长反应时间进行考察,发现随着时间的延长,其中相关杂质显著提高,同时由于温度的进一步降低,制冷成本将大幅上升,考虑到最终成品质量及成本问题,该反应优选反应温度-25 ℃。
2.4 考察结晶搅拌转速对反应收率的影响
具体实验数据如下表4:
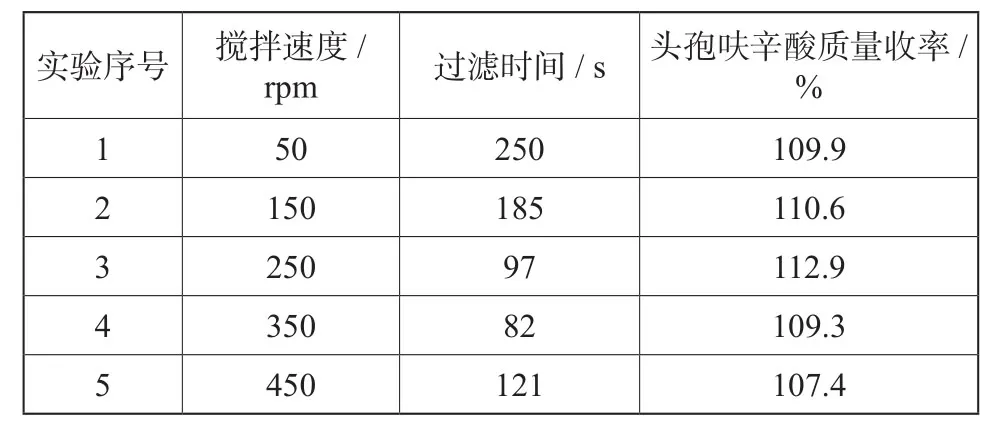
表4 结晶过程搅拌转速对收率的影响Tab. 4 Effect of different stirring speed for crystallization on the yield of product
由表4可知,实验3收率最高,且过滤时间也很短,实验1由于搅拌速度太慢,反应液体系中结晶不完全导致收率略低,同时由于搅拌太慢致使局部晶体爆出,形成无规则集聚体,致使过滤特别困难;试验5可能由于晶形太细,从而导致收率有所降低。搅拌对产品粒度分布影响较大。适当的搅拌速率可以给晶体提供更多的碰撞机会,并使溶液浓度保持均一,能够给晶体生长提供稳定的外部环境。但在搅拌过于激烈的情况下,随着颗粒的增大,晶体将承受更大的剪切力而被再次打碎小颗粒,不利于晶体稳定长大。
3 结论
本研究顺利地合成了头孢呋辛酸,首先,对酰氯化试剂三氯氧磷的用量进行了优化,该过程兼顾降低生产成本和对环境的保护理念,最终得出最优投料量,即n(SMIA):n(POCl3)为1∶1.2;其次,对DCC中间体制备过程中盐酸滴加时间进行优化,该步骤十分关键,盐酸滴加时间在影响质量的同时也会影响到产品收率,经过笔者大量实验最终得出最优滴加时间25 min;再次,对呋辛酸缩合反应时温度进行了优化,得最优反应温度-25 ℃;最后,对呋辛酸成品结晶过程做了优化,得出在搅拌转速为250 rpm时,生产操作容易进行且收率最高。
综上所述,本研究对头孢呋辛酸合成过程的关键点和参数进行了优化,使得中间体和成品收率分别为84.8 %和91.4 %,总收率高达77.5 %,该反应具有收率高 、低成本、易操作和环境友好等特点 ,并具有工业化生产价值,为生产头孢呋辛整个产业链奠定坚实的基础。
参考文献
[1]邓宝军,林诗贵,李蜀巍,等.头抱呋辛的临床抗感染研究进展[J].中国临床药理学杂志,2000,16(5):390-393.
[2]刘跃建,于云芝,李小惠,等.注射用头孢呋辛钠临床研究[J].中国抗生素杂志,2002,27(12):734-737.
[3]蔡仲曦,干荣富.我国头孢菌素抗生素之原料药与中间体现状与趋势分析[J].精细与专用化学品,2003,2:7-9,22.
[4]Bunnel C A. Process for Preparing Acid Halides:US,5084568[P].1992-01-28.
[5]Tsuji T,Okada T. Hydroxymethylcephem Compounds and Their Preparation:GB,218804-6[P]. 1987-09-23.
[6]李爱军,周雪琴,刘东志. 头孢呋辛钠合成工艺优化[J].天津大学学报,2007,40(11):1342-1345
[7]Siviero E,Cabri W,Terrassan D M. Process for the Preparation of β-lactam Derivatives:US,6458558[P].2002-10-01.
[8]Deshpande P B,Deshpande P N,Khadangale B P,et al. Process for the Preparation of Cef -uroxime Sodium:US,20040092735[P]. 2004-03-13.
[9]Kremminger P. Intermediates in Cephalosporin Production :US,20030171577[P]. 2003 -09-11.
[10]Humber D C,Laing S B,Weingarten G G,Process for the Preparation of Cephalosporin Compounds :US,4258183[P].1981-03-24.
[11]White H J,Eastlick D T,Oughton J F. Process for Preparing Sodium Cefuroxime:US,4775750[P].1988-10-04.
[12]Cabri W,Siviero E,Darverlo P L,et al. Process for the Synthesis of β-lactam Derivatives: US,6642378[P].2003-11-04.
[13]魏青杰,刘东,曹卫凯,等.头孢呋辛酸、头孢呋辛酯生产新技术开法[EB/OL].河北省科学技术研究成果公报第四号,河北省科学技术厅,2012,87-88. http://www.hebstd.gov.cn/ banshi/tongzhi/kejiting/content_79190.htm.
中图分类号:TQ 465
文献标识码:A
文章编号:2095-817X(2016)02-0035-000
收稿日期:2016-01-15
作者简介:曹卫凯(1987—),男,执业药师,主要从事药物合成和精细化工领域的研究。
Research of Synthesis of Cefuroxime
Cao Weikai
(Xian Wanlong Pharmaceutical Co., Ltd, Xi'an 710119)
Abstract:Cefuroxime was synthesized from 7-aminocephalosporin acid ( 7-ACA ). The synthesis process and the key points in the process were optimized. First, 7-ACA was hydrolyzed in the mixture of methanol and water under low temperature so as to produce D-7-ACA, which was not separated and acylation reaction was followed. Intermediate medium DCC was then yielded from crystallization and separation, the yielding rate of which may reach 84.8%. After the reaction of DCC and CSI to further finish carbamylation reaction, cefuroxime acid solution was obtained. With crystallization, drying and separation, the yielding rate of cefuroxime may reach 91.4%, so the total yielding rate is 77.5%. This process has the advantages of high yielding rate, low cost and easy operation, thus it is valuable in industrial production.
Keywords:cephalosporin; cefuroxime; synthesis; industrialization
