石墨电极中锂离子扩散的原位观测
2016-07-24朱建宇郭战胜冯捷敏戴文浩
朱建宇,郭战胜,2,冯捷敏,戴文浩
(1.上海市应用数学和力学研究所,上海200072;2.上海市力学在能源工程中的应用重点实验室,上海200072;3.上海大学理学院力学系,上海200444)
石墨电极中锂离子扩散的原位观测
朱建宇1,郭战胜1,2,冯捷敏2,3*,戴文浩3
(1.上海市应用数学和力学研究所,上海200072;2.上海市力学在能源工程中的应用重点实验室,上海200072;3.上海大学理学院力学系,上海200444)
锂离子在电极中的扩散机制是理解和研究锂离子电池各类性能的基础。设计了一个新型实验室模拟电池,实时原位观测了石墨电极充电过程中的颜色变化,研究了锂离子的扩散路径和机理。实验发现,锂离子从石墨电极边缘向中心进行扩散,边缘部分锂离子首先达到饱和状态。充电倍率显著影响锂离子扩散机理。充电倍率增大,LiC18相(对应于深蓝色区域)明显多于LiC12相(对应于红色区域),反映出锂离子浓度梯度增大。通过观测颜色变化,发现大倍率充电时可嵌入的锂离子少于小倍率。通过对石墨负极图像进行原位比色,可以估算出电极的实时充电状态。
锂离子电池;原位观测;锂离子扩散;原位比色
锂离子电池具有能量密度大、循环寿命长的优点,是目前和未来在混合动力汽车和纯电动汽车上非常有前景的能量储存装置。石墨由于安全性高、成本低、循环性能稳定,因此被用作许多商业锂离子电池的电极材料。不可避免的,锂离子电池存在容量衰减的问题[1-4],石墨电极中锂的不均匀分布是影响循环寿命的重要原因之一[5-7]。因而,不少学者对锂离子电池石墨电极进行了原位观测[8-18]。Eastwood等[8]用X射线断层摄影技术观察了锂离子电池石墨电极,低分辨可观察到超过1 000个颗粒组成的电极的孔隙率、表面体积比和孔弯曲率等,高分辨可分析单个粒子的表面体积比。Bulte等[9]通过电化学显微镜原位观察了石墨电极表面固液界面层(SEI膜)的变化。Honbo等[10]用拉曼光谱和扫描电子显微镜(SEM)研究了表面形貌不同的多种类石墨电极的电化学性能和锂沉积形貌。目前,对嵌锂时石墨电极颜色变化的研究不多,主要集中在锂/石墨结构的半电池中[11-15]。Maire等[11-12]介绍了原位比色法作为一种快速、低成本的方法测量各种锂离子电池电极中的锂含量。同时,还通过实验得到了多孔石墨电极的表观扩散系数和活化能。Harris等[13-14]通过观察石墨颜色变化发现石墨电极中锂化边界的移动受液相扩散的控制。此外,他们还通过对石墨颜色变化图像的分析得到了相应的电极变形和应变场[15]。室温下,石墨的嵌锂过程会形成多种锂-石墨层间化合物[16-18]:LiC72、LiC36、LiC27、LiC18、LiC12和LiC6。有些锂-石墨层间化合物具有区别于原始灰黑色的独特颜色,如LiC18、LiC12和LiC6,分别为深蓝色、红色和金色。同样的,针对商业全电池石墨电极的实时原位观测也非常重要,但是这方面的研究非常少。
充电状态(State of Charge,SOC)是用来描述电池使用过程中可充入和放出容量的重要参数,其数值定义为电池剩余容量与电池容量的比值[19]。目前,对电池SOC的估算方法主要有:放电实验法、电量累积法、开路电压法、人工神经网络法、卡尔曼滤波法等[20-24]。放电实验法[20]具有可靠,精度高的优点,缺点是需要较长时间,测量时电池必须处于脱机状态。电量累积法[21]是一种简单、可靠的估计方法,缺点是由于电流测量精度的原因会导致累计误差。开路电压法[23]简单易行,精度较高,缺点是电池组需要静置较长时间达到稳定状态,以克服自恢复效应。卡尔曼滤波法[24]不仅能够获得SOC的估计值,还能得到其估计误差,缺点是需要建立准确的电池模型,运算量较大,能力要求高。人工神经网络法[22]快速、方便,具有较高精度,可以由现场工况来确定电池的SOC,缺点是估计误差受数据和训练方法的影响大,而且需要大量的训练数据。如何直观简单地计算全电池实时的SOC对于研究锂离子电池性能和寿命是非常重要的。
本文设计了一个新型实验室模拟电池,宏微观地实时原位观测了石墨/磷酸铁锂电池中石墨负极在嵌锂过程的颜色变化。根据捕捉到的原位图像分析了锂离子在石墨负极中的扩散形式,研究了不同充电倍率对锂离子扩散的影响。利用原位比色法估算出了石墨负极的实时SOC值。该方法具有实时、直观以及计算方便的优点。
1 实验
1.1 实验材料和充放电规程
实验使用95.7%的石墨作为负极活性材料,粘合剂为SBR+CMC,集流体为铜箔;正极活性材料使用LiFePO4,集流体为铝箔。LiFePO4正极和石墨负极厚度均是100 mm。隔膜为Celgard 2325三层隔膜,厚度为25 mm。电解液使用1 mol/L LiPF6/(EC+DMC)。常温下(25±1)℃,在充满氩气的手套箱中组装成石墨/隔膜/LiFePO4全电池。
1.2 实时原位观测的模拟电池结构
如图1所示,为了便于实时观测并且不破坏电池结构,实验中采用边对边的组装方式,而非商用缠绕或叠片结构的面对面方式,LiFePO4正极被隔膜完全包覆,与石墨负极边对边排列,正、负极自由端的间距为1~2 mm。实验室模拟电池使用的边对边结构能够确保在充电过程中实时地捕捉石墨电极的颜色变化。石墨活性层面积为4 mm×8 mm,容量为1.267 2 mAh(面密度120 g/m2,理论比容量330 mAh/g),为了保证Li+充足,LiFePO4活性层面积4 mm×30 mm,容量为3.264 mAh(面密度160 g/m2,理论比容量为170 mAh/g)。
1.3 充放电规程与测试方法
实验使用BK-6808电池充放电仪以不同倍率(0.2C、0.5C和1C)恒流对模拟电池进行充放电,上下限电压为2.0~4.2 V。充、放电结束后保持开路状态5 min,数据收集频率为1 Hz。使用数码显微镜对石墨负极的颜色进行原位实时拍摄,图像采集时间间隔为每30 s一次。
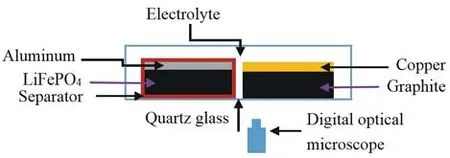
图1 电池组装结构示意图
2 结果与讨论
2.1 石墨电极变色现象
石墨具有层状结构,其嵌锂的过程是锂离子嵌入石墨层间的过程。锂离子嵌入后,层面保持平面,石墨层与嵌入层平行排列,而且是每隔一层、二层、三层…有规则地嵌入,分别称为一阶、二阶、三阶…石墨层间化合物[25]。其中,具有独特颜色的石墨层间化合物的反应可表示为[17-18]:

图2为不同石墨层间化合物的颜色。在室温下,未嵌入锂离子的石墨呈现灰黑色,如图2(a)所示。锂离子隔两个石墨层嵌入产生稀释Ⅱ阶LiC18相,LiC18相中锂原子排列不规则,此时石墨电极颜色为深蓝色,如图2(b)。锂离子继续嵌入,随着LiC18相的消失LiC12相生成,锂原子排列逐渐规则化,稀释Ⅱ阶LiC18相完全转化为Ⅱ阶LiC12相,此时石墨电极颜色为红色,如图2(c)。当LiC12相饱和,锂离子将嵌入到未被占用的石墨层中,在LiC12相消失的同时生成规则排列的Ⅰ阶LiC6相,此时石墨电极颜色为金色,如图2(d)。本文利用这几个相的颜色变化研究锂离子在石墨电极中的扩散方式。

图2 不同石墨层间化合物的颜色
石墨负极的尺寸为8 mm(长边)×4 mm(短边)×0.091 mm(厚度),短边与厚度之比为44∶1,所以本文认为石墨厚度方向锂离子浓度均匀,可以忽略锂离子在厚度方向的扩散。图3是在0.2C恒流充电下捕捉到的石墨电极实时原位图像。从图像来看,石墨上端与下端对称、左端与右端不对称,这是由于电池正负极几何结构是边对边形式导致的。石墨负极右端靠近磷酸铁锂正极,充电时电极右端的锂离子浓度会高于电极左端,所以电极右端更容易嵌入锂离子,产生左端与右端不对称的现象。图3(a)是石墨负极的初始状态,此时无锂嵌入,电极呈灰黑色。图3(b)显示,由于锂离子的嵌入,灰黑色石墨负极逐渐被深蓝色、红色和金色由四边向中心取代。电极右端最外层为金色,向内依次为红色、深蓝色和灰黑色。随着锂离子的继续嵌入,图3(c)中电极左端由边缘向中心方向出现了金色和红色,与右端相比有明显的滞后性,但颜色变化趋势一致。此外,电极中心部位初始的灰黑色消失,被深蓝色完全替代。当充电到第194 min,如图3(d)所示,深蓝色完全消失,电极中心完全转化为红色,且金色内边界亦不断向中心移动。图3(e)中,金色内边界从边缘向中心逐渐移动,红色外围已经转化为了金色。充电结束时,红色也全部消失,整个石墨负极完全转化为了金色,如图3(f)所示。图中气泡的产生是由于石墨负极和电解液的界面反应形成SEI膜导致,主要成分为CO2,C2H4等气体[26-27]。

图3 0.2C倍率充电过程中石墨负极的实时原位图像
图4更清晰地演示了嵌锂时锂离子在石墨负极中的扩散方式。图4(a)、(b)和(c)分别是石墨电极充电到第74、122和194 min时的图像。图中的1、2、3和4分别对应于灰黑色、深蓝色、红色和金色四个颜色区域,箭头上的分割线表示颜色边界。由前述锂化反应和各锂-石墨层间化合物的颜色可知,4代表的金色是由石墨完全锂化为锂-石墨层间化合物LiC6所产生的变化,是锂浓度最高的区域。同理,2和3代表的深蓝色和红色也是由石墨分别锂化为LiC18和LiC12产生,根据石墨的锂化程度可以知道3处的锂浓度大于2。1代表的灰黑色暂时无锂嵌入,是锂浓度最低的区域。因此,图中箭头的方向代表了锂浓度降的方向,也是锂离子在石墨中扩散的方向。实验表明,在充电时锂离子先在石墨负极边缘集中,与边缘的石墨先形成锂碳化合物,然后再逐步向中间扩散。

图4 石墨负极充电过程中的颜色图像
2.2 充电倍率对锂离子扩散的影响
图5是相同的三个电池在其他条件相同的情况下,以不同的充电倍率0.2C、0.5C和1C充电到SOC为50%(电量累计法计算所得)时的图像。可以看到三种倍率下,石墨负极均呈现出深蓝色、红色和金色共存,灰黑色消失的状态。其中,深蓝色、红色和金色三种颜色的面积占总面积之比在0.2C倍率下分别是23%、53%和24%;0.5C倍率下是46%、28%和26%;1C倍率下是44%、33%和23%。从这三组数据可以发现,用小倍率0.2C充电时,红色LiC12相最多,深蓝色LiC18相最少。增大充电倍率后,红色LiC12相减少,深蓝色LiC18相增多,金色LiC6相不变。倍率增大后深蓝色LiC18相明显多于红色LiC12相,表明锂离子浓度梯度增大。这种情况是由于充电倍率增大之后,锂离子的扩散浓度增大,在锂离子的扩散路径上发生锂化反应时锂往往都是富足的一方,所以红色LiC12相生成后会继续被锂化直至生成金色LiC6相。显然这种情况只发生在靠近边缘的地方,这可能是由于扩散浓度增大的同时会造成扩散路径的堵塞,阻碍锂离子向中心传输,因此对于中心部位的锂化反应,锂离子是不足的,导致了中心部位深蓝色LiC18相增多。
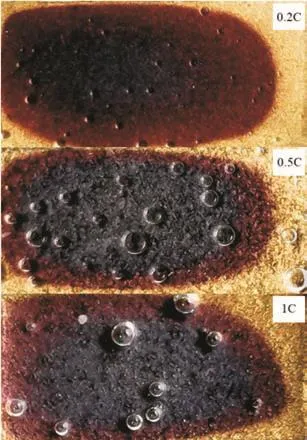
图5 不同充电倍率下SOC达到50%时石墨电极的图像
图6是三种充电倍率下电压-时间曲线。由电压-时间曲线可以明显地看出,0.2C倍率充电时,电压曲线可分为三段:a、b和c,如图所示。a段,电压增大且增大速率逐渐变缓;b段,出现电压平台;c段,电压增大且增大速率逐渐变快;0.5C倍率充电时,a段段末电压比0.2C大,并且没有电压平台,电压曲线直接进入c段;1C倍率充电时,a段的段末电压最大,同样没有电压平台。图7是充电结束时石墨负极的图像。根据充电结束时石墨负极的颜色,能够清晰地判断出1C倍率下电极的锂化程度最小,0.2C倍率下最大。电量累计法计算出不同充电倍率下的截止SOC分别为86%(0.2C)、75%(0.5C)和56%(1C)。

图6 不同充电倍率的电压-时间曲线图
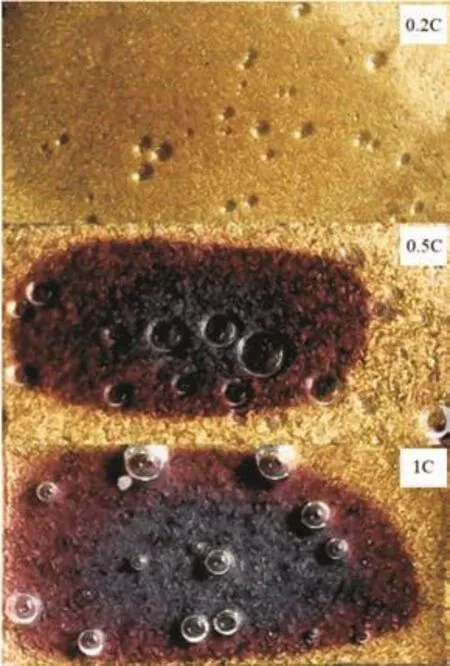
图7 充电结束时的石墨电极图像
可以发现,在相同的条件下进行恒流充电,小倍率充电的电池,可充入较多的电量。从电压曲线可知,没有电压平台存在的情况下可充入的电量明显减少。此外,同样没有电压平台的1C倍率和0.5C倍率相比,1C倍率下可充入的电量比0.5C更少,这和1C倍率下a段段末电压较大有关。a段段末电压大,那么达到截止电压所需时间会变短,可充入的电量必然减少。大倍率充电时,a段段末电压高以及b段电压平台消失有可能是电极极化[28]造成的。极化的程度可用超电势来表示,超电势是指某一电流密度下,电极电势与平衡电极电势的差值。在所有极化超电势中,浓差超电势和电化学超电势的影响最大。一般来说,充电倍率越大,电流密度就越大,那么超电势越大,从而造成电压虚高,平台期消失,最终导致可充入的电量减少。
2.3 原位比色方法估算SOC
SOC除了可以用容量定义外,也可以用石墨中锂含量的多少来进行计算。石墨中实时锂含量与最大锂含量的比值即为当前的SOC值。从化学成份上分析不同锂-石墨层间化合物,能够得到不同锂化程度的石墨电极所对应的SOC状态。金色LiC6相出现时表示石墨已经被完全锂化,锂的含量达到饱和,所以对应的SOC状态是100%,而此时C和Li的原子比为6∶1。根据红色LiC12相中C和Li的原子比为12∶1可知,红色LiC12相中的锂含量是金色LiC6相的一半,因此同一电极中红色LiC12相对应的SOC状态是50%。同理,深蓝色LiC18相对应的SOC状态是33%。
通过图8所示,可以计算得到石墨负极图像上不同颜色区域面积与总面积之比。结合前述不同相对应的SOC状态,可以推算出石墨负极根据原位比色方法估算的SOC,如式(4)所示:

式中:SD、SR和SG分别表示深蓝色相、红色相和金色相面积与电极总面积之比。图9为原位比色方法和电量累计法的SOC估算值。由于电流恒定,所以0.2C充电时电量累计法估算的SOC-时间t曲线是线性的(黑色线段)。原位比色方法的SOC估算值与电量累计法的SOC估算值比较接近,相差在10%左右。运用原位比色能够实时原位估算石墨负极的SOC状态,比目前现有的方法更加直观。

图8 石墨电极上不同颜色面积比重

图9 原位比色方法和电量累计法估算SOC
3 结论
本文设计了宏微观实验室模拟电池,实时原位观测了充电过程中石墨负极的颜色变化以及锂离子在石墨负极中的扩散。充电倍率直接影响锂离子在电极中的扩散,充电倍率增大,红色LiC12相的含量减小、深蓝色LiC18相增多,锂离子浓度梯度增大。此外,大倍率充电时可充入的电量明显少于小倍率,这是由于电极极化导致的电压虚高造成的。通过对电极图像原位比色,估算出了电极实时SOC,这种方法具有实时、直观的优势。
[1]BROUSSELY M,BIENSAN P H,BONHOMME F,et al.Main aging mechanisms in Li ion batteries[J].J Power Sources,2005,146:90-96.
[2]ARORAT P,WHITE R E.Capacity fade mechanisms and side reactions in lithium-ion batteries[J].J Electrochem Soc,1998,145(10):3647-3667.
[3]AURBACH D.A review on new solutions,new measurements procedures and new materials for rechargeable Li batteries[J].J PowerSources,2005,146:71-78.
[4]VETTER J,NOV魣K P,WAGNER M R,et al.Ageing mechanisms in lithium-ion batteries[J].J Power Sources,2005,147:269-281.
[5]MIGGE S,SANDMANN G,RAHNER D,et al.Studying lithium intercalation into graphite particles via in situ Raman spectroscopy and confocal microscopy[J].J Solid State Electrochem,2005,9:132-137.
[6]HARDWICK L J,BUQA H,NOV魣K P,et al.Graphite surface disorder detection using in situ Raman microscopy[J].Solid State Ionics,2006,177:2801-2806.
[7]REYNIER Y,YAZAMI R,FULTZ B.XRD evidence of macroscopic composition inhomogeneities in the graphite-lithium electrode[J].J Power Sources,2007,165:616-619.
[8]EASTWOOD D S,BRADLEY R S,TARIQ F,et al.The application of phase contrast X-ray techniques for imaging Li-ion battery electrodes[J].Nuclear Instruments and Methods in Physics Research B,2014,324:118-123.
[9]B譈LTER H,PETERS F,SCHWENZEL J,et al.Spatiotemporal changes of the solid electrolyte interphase in lithium-ion batteries detected by scanning electrochemical microscopy[J].Angew Chem Int Ed,2014,53:10531-10535.
[10]HONBO H,TAKEI K,ISHII Y,et al.Electrochemical properties and Li deposition morphologies of surface modified graphite after grinding[J].J Power Sources,2009,189:337-343.
[11]MAIRE P,KAISER H,SCHEIFELE W,et al.Colorimetric determination of lithium-ion mobility in graphite composite electrodes[J].J Electroanal Chem,2010,644:127-131.
[12]MAIRE P,EVANS A,KAISER H,et al.Colorimetric determination of lithium content in electrodes of lithium-ion batteries[J].J Electrochem Soc,2008,155(11):A862-A865.
[13]HARRIS S J,TIMMONS A,BAKER D R,et al.Direct in situ measurements of Li transport in Li-ion battery negative electrodes[J].Chem Phys Lett,2010,485:265-274.
[14]HARRIS S J,RAHANI E K,SHENOY V B.Direct In Situ observation and numerical simulations of non-shrinking-core behavior in an MCMB graphite composite electrode[J].J Electrochem Soc,2012,159(9):A1501-A1507.
[15]QI Y,HARRIS S J.In Situ observation of strains during lithiation of a graphite electrode[J].J Electrochem Soc,2010,157(6):A741-A747.
[16]AURBACHA D,MARKOVSKY B,WEISSMAN I,et al.On the correlation between surface chemistry andperformance of graphite negative electrodes for Li ion[J].Electrochim Acta,1999,45:67-86.
[17]DAHN J R,ZHENG T,LIU Y H,et al.Mechanisms for lithium insertion in carbonaceous materials[J].Science,1995,270:590-593.
[18]DOLL G L,EKLUND P C.Raman scattering study of the high-frequency graphitic intralayer modes in Li-graphite and the stage dependence of the mode frequency in donor graphite intercalation compounds[J].Phys Rev B,1987,36(9):4940-4945.
[19]安志胜.电池管理系统中锂离子电池SOC估算方法的研究[D].太原:太原科技大学,2013.
[20]孙骏,李宝辉,薛敏.电动汽车SOC估算方法[J].汽车工程师,2011,12:25-27.
[21]林成涛,王军平,陈全世.电动汽车SOC估计方法原理与应用[J].电池,2004,34(5):376-378.
[22]CHAN C C,LO E W C,SHEN W X.The available capacity computation model based on artificial neural network for lead-acid batteries in electric vehicles[J].J Power Sources,2000,87:201-204.
[23]HUET F.A review of impedance measurements for determination of the state-of-charge or state-of-health of secondary batteries[J].J Power Sources,1998,70(1):62-68.
[24]文锋,林程,姜久春,等.电动汽车用锂离子电池荷电状态定义和估算方法[J].高技术通讯,2012,22(9):975-979.
[25]简志敏.锂离子电池用扩层石墨负极材料的研究[D].长沙:湖南大学,2012.
[26]DEDRYV魬RE R,MARTINEZ H,LEROY S,et al.Surface film formation on electrodes in a LiCoO2/graphite cell:A step by step XPS study[J].J Power Sources,2007,174:462-468.
[27]AURBACH D,MASHKOVICH M.A study of lithium deposition-dissolution processes in a few selected electrolyte solutions by electrochemical quartz crystal microbalance[J].J Electrochem Soc,1998,145(8):A2629.
[28]郭鹤桐,覃奇贤.电化学教程[M].天津:天津大学出版社,2000:325-328.
In situ observation of lithium ion diffusion in graphite electrode
ZHU Jian-yu1,GUO Zhan-sheng1,2,FENG Jie-min2,3*,DAI Wen-hao3
(1.Shanghai Institute of Applied Mathematics and Mechanics,Shanghai 200072,China;2.Shanghai Key Laboratory of Mechanics in Energy
Engineering,Shanghai 200072,China;3.Department of Mechanics,College of Science,Shanghai University,Shanghai 200444,China)
Diffusion mechanism of lithium ion in the electrode is the basis to understand and research all kinds of lithium ion battery properties.The new experimental device was designed to realize the real time in situ observation of the color change in graphite electrode during charging.The path and mechanism of lithium ion diffusion were researched.It is found that lithium ions diffuse from the edge to the center of graphite electrode.The edge portion lithium ion reaches saturation firstly.Charging rate significantly affects the diffusion mechanism of lithium ion.LiC18phase(corresponding to the dark blue region)is more than LiC12phase(corresponding to the red region)at high charging rate,indicating that lithium ion concentration gradient is greater.By observing color change,It is found that lithium ion enable to inset into electrode at high rate is less than that at low rate.In situ colorimetry of the graphite electrode images can estimate the real time state of charge of the electrode.
lithium ion battery;in situ observation;lithium ion diffusion;in situ colorimetry
TM 912
A
1002-087 X(2016)08-1550-04
2016-01-17
国家自然科学基金(11472165、11332005);上海市自然科学基金(12ZR1410200)
朱建宇(1991—),男,江苏省人,硕士生,主要研究方向为锂电池电极材料的力学性能。
冯捷敏,Email:fengjiemin@shu.edu.cn
