紫外多波长光谱法监测阿司匹林合成过程的水杨酸和阿司匹林
2016-07-20张祖栋孙阔孙浩
张祖栋,孙阔,孙浩
(廊坊市环境监测站,河北廊坊 065000)
紫外多波长光谱法监测阿司匹林合成过程的水杨酸和阿司匹林
张祖栋,孙阔,孙浩
(廊坊市环境监测站,河北廊坊 065000)
摘要建立测定阿司匹林合成过程中水杨酸、阿司匹林含量和转化率的分析模型。采用紫外多波长扫描乙醇溶液中阿司匹林和水杨酸的紫外光谱,建立水杨酸和阿司匹林紫外光谱的向量长度与其质量浓度的标准曲线,通过斜投影算法分离出待测样本中阿司匹林和水杨酸的紫外光谱。计算光谱向量长度,代入标准曲线得到待测样本中阿司匹林和水杨酸的含量。实验结果表明,水杨酸、阿司匹林的质量浓度与其紫外光谱的向量长度呈良好的线性关系,线性范围分别为2.00~40.00,10.00~200.00 μg/mL。水杨酸和阿司匹林测定结果的相对标准偏差为0.16%~2.19%(n=5),加标回收率在93.3%~106.9%之间。该方法快速简便、准确可靠,可满足阿司匹林和水杨酸的同时测定及反应过程中水杨酸转化率监测要求。
关键词紫外多波长;向量长度;斜投影;水杨酸;阿司匹林
阿司匹林合成过程中水杨酸转化率的测定不但可以有效提高阿司匹林生产效率、减少生产成本,而且对阿司匹林生产质量控制有着重要意义。传统的分析方法如滴定[1]、高效液相法[2-3]等分析时间长,步骤繁琐。
近年来随着化学计量学方法[4-5]的发展,多元信号的分辨与校正成为分析化学的研究关键,多元校正方法所选的波长不必对分析物有选择性[6-8],也可以对样本中的其它化学物质有响应,专利[9]公开了利用子空间角判据实现多组分体系中的组分辨识,它采用张量空间描述多组分体系的光谱,每一种物质的光谱可用空间中的一个向量表示,向量的长度(即模量)代表物质的含量。据此笔者建立水杨酸和阿司匹林紫外光谱的向量长度与浓度的关系,通过扫描阿司匹林合成过程中样本乙醇溶液的多波长紫外光谱,以斜投影算法[10-13]分离出样本中阿司匹林和水杨酸的紫外光谱图,以此测定氨基磺酸催化合成阿司匹林体系中水杨酸、阿司匹林的含量和转化率。
1 原理及计算方法
斜投影算法以数据空间描述多变量体系,是一种多变量体系中纯信号处理的分析方法。把数据空间M划分为被测变量的向量张成的子空间S和其余变量向量张成的相邻空间H,以向量S和H互为关系建立纯信号分离模型,将变量S从M中分离出来,斜投影算子E按式(1)计算[14]。
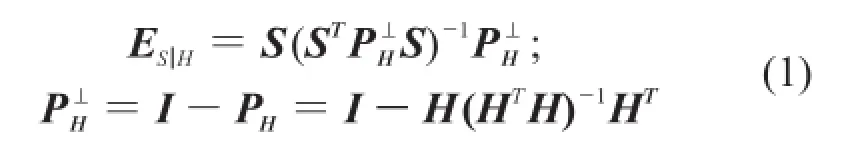
式中:T——矩阵转置;
I——与PH维数相同的单位矩阵。
对光谱求向量长度按式(2)计算。
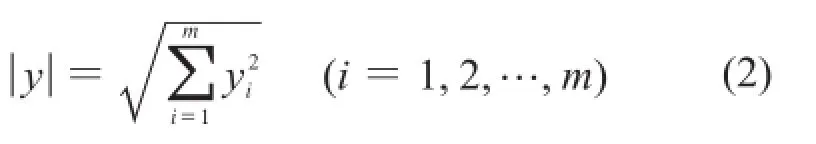
式中:i——波长序号;
yi——第i个波长下的吸光度。
2 实验部分
2.1 主要仪器与试剂
光纤光谱仪:Maya2000Pro型,美国海洋光学公司;
艾科勒分析天平:ALC-210.4型,德国赛多利斯集团公司;
水杨酸、乙酰水杨酸、乙酸酐、氨基磺酸、无水乙醇:分析纯。
2.2 标准样品及光谱库
以下均采用光程为1 cm石英比色皿,以无水乙醇为参比,采集样品在250~345 nm波长范围内的紫外光谱。以无水乙醇为溶剂,在2.00~40.00 μg/mL浓度范围内,采集浓度为2,5,10,15,20,30,40 μg/mL水杨酸标准溶液的紫外光谱;在10.00~200.00 μg/mL浓度范围内,采集浓度为10,20,40,80,120,160,200 μg/mL的阿司匹林标准溶液的紫外光谱;采集乙酸、乙酸酐的紫外光谱。
2.3 阿司匹林的合成与过程监测
按文献方法[8],磁力搅拌下将20.73 g乙酸酐于50 mL三口烧瓶中,水浴温度升至81℃,依次加入13.88 g水杨酸和0.25 g氨基磺酸,恒温反应18 min。每间隔1 min取样1 mL于100 mL容量瓶中并用无水乙醇定容;再取出2 mL以无水乙醇定容于50 mL容量瓶,作为待测样品N1,N2,…,N18。
2.4 数据处理
2.4.1 水杨酸定量过程
(1)对被测组分水杨酸的乙醇溶液的系列浓度xj={x1,x2,…,xn}的紫外光谱yj求模长经最小二乘回归后得标准曲线yj=ajxj+bj。
(2)测定阿司匹林、乙酸酐、乙酸、氨基磺酸的乙醇溶液的紫外光谱,作为背景光谱n。
(3)测量待测混合物样品乙醇溶液的紫外光谱。
(4)根据斜投影算子从待测混合物样品乙醇溶液紫外光谱中分离出水杨酸的纯光谱y'求模长y'代入标准曲线得出水杨酸浓度。
2.4.2 阿司匹林定量过程
将上述过程中的水杨酸替换成阿司匹林重复上述步骤,计算阿司匹林样品浓度。
以上数据处理过程均在MATLAB7.8.0中实现。
3 结果与讨论
3.1 体系中各组分的紫外吸收光谱
在无水乙醇中,水杨酸、乙酸酐、氨基磺酸和乙酰水杨酸在250~345 nm波长范围内的紫外吸收光谱见图1。由图1可知,阿司匹林和水杨酸最大吸收波长分别为275.18 nm和304.21 nm,氨基磺酸在此波长范围内无紫外吸收,水杨酸、阿司匹林、乙酸酐和乙酸4组分光谱有重叠。
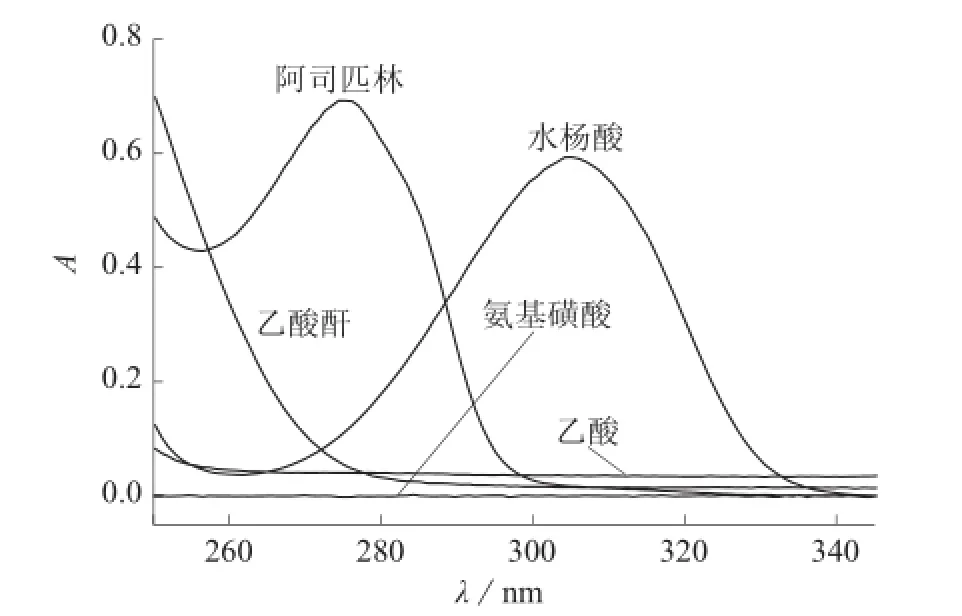
图1 反应体系中各组分紫外吸收光谱
3.2 线性方程
水杨酸的紫外光谱和阿司匹林的紫外光谱模长(y)与其对应的水杨酸、阿司匹林标准溶液浓度(x)进行最小二乘拟合,得到线性回归方程分别为y =0.027 1x-0.000 8,(r=0.999 6);y =0.005 5x+0.018 8,r=0.999 3。结果表明,水杨酸、阿司匹林的质量浓度分别在2.00~40.00 μg/mL 和10.00~200.00 μg/mL范围内与其光谱模长具有良好的线性关系,线性方程分别见图4、图5。
3.3 模型对合成体系样品测定
阿司匹林合成过程样品紫外光谱及被分离出水杨酸和阿司匹林的紫外光谱分别见图2、图3。
分别对被分离出的水杨酸和阿司匹林的紫外光谱向量求模长,将求得的模长代入水杨酸和阿司匹林的线性方程,所得的样品分析结果分别见图4、图5。

图2 阿司匹林合成过程中样品紫外吸收光谱

图3 阿司匹林合成过程样品分离光谱
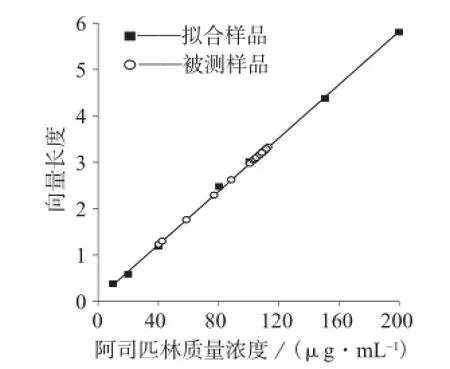
图4 阿司匹林分析曲线及样品点分布

图5 水杨酸分析曲线及样品点分布
将上述计算结果转化成水杨酸的转化率,其反应过程变化及趋势如图6所示。由图6可知,氨基磺酸催化反应在81℃合成阿司匹林的反应过程前期反应剧烈,水杨酸转化率变化较明显,8 min后反应较为缓和逐渐达到平衡,平衡时水杨酸的转化率达到98.0%。

图6 合成过程中水杨酸转化率变化趋势
3.4 精密度试验
选择反应2,4,8 min时对应的3个样品,分别进行5次平行测定,结果见表1。

表1 精密度试验结果
由表1可知,水杨酸和阿司匹林测定结果的相对标准偏差不大于2.19%,说明该方法的精密度良好,此分析模型满足检测要求。
3.5 加标回收试验
选用反应过程的9个样品,在标准曲线浓度范围内,加入一定量水杨酸和阿司匹林定容至50 mL,计算水杨酸和阿司匹林的回收率,结果见表2。由表2可知,阿司匹林和水杨酸的回收率在93.3%~106.9%之间,表明该模型具有良好的准确度,可定量分析氨基磺酸催化合成阿司匹林过程体系中的水杨酸和阿司匹林。
4 结论
采用紫外多波长监测法扫描乙醇溶液中阿司匹林和水杨酸的紫外光谱,计算水杨酸和阿司匹林紫外光谱的向量长度与浓度定量关系,建立测定阿司匹林合成过程中水杨酸、阿司匹林含量和转化率的分析模型。通过斜投影算法分离出待测样本中阿司匹林和水杨酸的紫外光谱,计算光谱向量长度,代入标准曲线可求待测样本中阿司匹林和水杨酸含量。该方法快速简便、准确可靠,可满足阿司匹林和水杨酸的同时测定及反应过程水杨酸转化率监测。
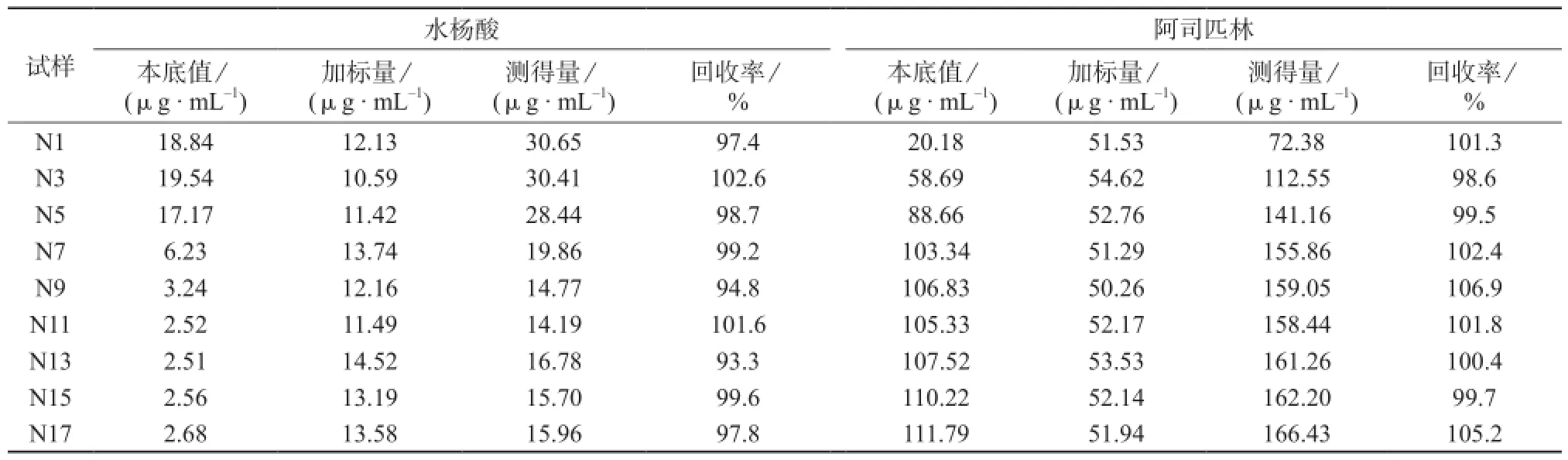
表2 水杨酸和阿司匹林的加标回收试验结果
参 考 文 献
[1] 国家药典委员会.中华人民共和国药典[M].2部.北京:化学工业出版社,2010: 284.
[2] 李克庆.测定阿司匹林肠溶衣片中阿司匹林含量及游离水杨酸[J].中国药师,2006,9(10): 925-927.
[3] Hiral J Panchal,Bhanubhai N Suhagia,Natvarlal J Patel,et al. Simultaneous estimation of atorvastatin calcium,ramipril and aspirin in capsule dosage form by RP-LC[J]. Chromatographia,2009,69: 91-95.
[4] 倪永年.化学计量学在分析化学中的应用[M].北京:科学出版社,2004: 161-178.
[5] 梁逸曾.白灰黑复杂多组分分析体系及其化学计量学算法[M].长沙: 湖南科学技术出版社,1996: 26-36.
[6] Sena M M, Poppi R J. N-way PLS applied to simultaneous spectrophotometric determination of acetylsalicylic acid,paracetamol and caffeine[J]. Journal of Pharmaceutical and Biomedical Analysis,2004,34(1): 27-34.
[7] 王桂芳,李光和,窦英,等.偏最小二乘法测定复方乙酰水杨酸片中的有效成分[J].化学分析计量,2005,14(1): 40-42.
[8] Mot A C, Soponar F, Medvedovici A, et al. Simultaneous spectrophotometric determination of aspirin, paracetamol, caffeine,and chlorphenamine from pharmaceutical formulations using multivariate regression methods[J]. Analytical Letters,2010,43(5): 804-813.
[9] 姚志湘,粟晖.基于角度度量的多变量分析方法:中国专利,201110188187 [P]. 2012-01-04.
[10] 张伟,李克新,武春风,等.多光谱目标探测的波段选择[J].光学技术,2005,31(6): 893-897.
[11] 葛军,姚志湘,粟晖,等.基于斜投影算法对混合染液不展开多波长薄层色谱定量分析[J].印染助剂,2013,30(10): 47-50.
[12] 张小飞,徐大专.基于斜投影的波束成型算法[J].电子与信息学报,2008,30(3): 585-588.
[13] 杨树.氨基磺酸催化合成乙酰水杨酸的研究[J].昆明师范高等专科学校学报,2007,29(4): 108-109.
[14] 张贤达.矩阵分析与应用[M].北京:清华大学出版社,2001:55-59.
联系人:孙阔;E-mail: sunkuo2009@163.com
中图分类号:O657.3
文献标识码:A
文章编号:1008-6145(2016)01-0030-04
doi:10.3969/j.issn.1008-6145.2016.01.008
收稿日期:2015-10-20
Monitoring Salicylic Acid and Aspirin in Synthesis Systems of Aspirin by Muti-Wavelength Ultraviolet Spectroscopy
Zhang Zudong, Sun Kuo, Sun Hao
(Langfang City Environment Monitoring Center, Langfang 065000, China)
AbstractIn order to establish the analysis model for the quantitative determination of salicylic acid and aspirin in synthesis systems of aspirin, a method based on quantitative relation between length of ultraviolet spectroscopy vector and concentration was developed. Pure spectrum of salicylic acid and aspirin in testing sample were separated through oblique projection method. The experimental results showed that the mass concentration of salicylic acid, aspirin and its ultraviolet spectral vector length had good linear relationship in the range of 2.00-40.00, 10.00-200.00 μg/mL, respectively. The relative standard deviation of determination results was 0.16%-2.19%(n=5), and the recoveries ranged from 93.3% to 106.9% for salicylic acid and aspirin. The method is quick and simple, accurate and reliable, so it can meet the requirments of simultaneous determination of aspirin, salicylic acid and salicylic acid conversion in reaction process monitoring.
Keywordsmuti-wavelength; vector length; oblique projection; salicylic acid; aspirin
