基于互信息的显著基因提取及转录调控网络构建
2016-07-19牟晓阳
孔 薇 支 星 牟晓阳
1(上海海事大学信息工程学院 上海 201306)2(罗文大学生物化学系 新泽西 08028 美国)
基于互信息的显著基因提取及转录调控网络构建
孔薇1支星1牟晓阳2
1(上海海事大学信息工程学院上海 201306)2(罗文大学生物化学系新泽西 08028 美国)
摘要传统特征基因提取方法往往只注重单个基因在不同样本中的表达差异,忽视了基因之间的关联性以及多个致病基因作为一个基因模块与复杂疾病的联系。针对这种情况,提出基于互信息MI(Mutual Information)的特征基因提取算法,提取在健康对照和阿尔茨海默症AD(Alzheimer’s disease)患病样本中关联度具有明显差异变化的基因作为特征基因。在此基础上,结合转录因子TF(Transcription factor)对靶基因TG(Target gene)调控的生物学先验信息,利用网络成分分析NCA(Network Component Analysis)算法分析转录因子的表达活性及其对靶基因的调控强度,构建AD特征基因的转录调控网络。分子生物学分析表明,它们在有丝分裂、细胞周期、免疫反应以及炎症反应过程中的变化对AD的退化过程具有重要作用。
关键词阿尔茨海默症(AD)互信息(MI)转录因子(TF)网络成分分析(NCA)
0引言
阿尔茨海默症[1]AD是以进行性认知障碍和记忆力损害为主的中枢神经系统退行性病变。在过去的几十年中,虽然已经提出了多种假说和许多公认的AD易感基因,但是AD的遗传学机制和发病机制仍不清楚。对于探寻基因表达水平,转录因子TF活性以及转录调控机制变化,表现出的转录因子和靶基因TG的协调动态响应,将对于研究AD的致病机理具有重大意义。
近年来,随着高通量技术的飞速发展,如DNA微阵列技术和统计学计算工具[2]使得生物信号大规模的测量,发现了AD有关的许多重要基因、共调控基因群以及转录调控网络。此外,为了克服微阵列技术的基本缺点,如小样本、基因数量大、测量误差以及信息不全。一些其他的高通量技术,如蛋白质相互作用数据PPI(protein-proteininteraction)、转录因子和microRNA信息数据也相继被整合,使得对于AD致病机理的研究有了更加充分的信息数据量。传统的基因网络分析模型如独立成分分析[3]ICA(independentcomponentanalysis)并不能运用一些转录调控信息,并且只是纯粹的基于调控信号的数学和统计特性,因此它们并不能准确地构建转录调控网络。网络成分分析[4]NCA是一种用来确定转录因子对靶基因调控影响和其活性强度的方法,算法思想是通过预先输入基因表达数据和先验给定的转录因子和其靶基因的连接信息,将基因表达数据转化成TF活性水平和转录因子对其靶基因的调控影响。NCA已被用于许多研究,例如,应用于酵母细胞周期过程中,识别此前被忽视的振荡活性模式[4]。
传统的差异基因提取方法,往往是基于单条基因在健康对照和患病样本中的表达水平差异,表达差异的大小表明了该基因与疾病的相关性大小。但是却忽视了基因之间的关联性以及多个致病基因作为一个基因模块与复杂疾病的相关性。张焕萍等人基于互信息(MI)和最大团(clique)相结合的方法,挖掘出有关结肠癌(Colon)的差异共表达致病基因模块[5]。本文从基因之间的关联性角度出发,基于互信息方法,提取出在健康组样本中关联度低甚至没有关联性,而在患病样本中关联度高的基因,并将它们作为特征基因。该算法旨在通过计算任意两条基因表达谱在两类不同样本中的互信息值,得出对应的互信息矩阵,然后围绕这两个互信息矩阵去构建一个图的邻接矩阵,最终通过这个邻接矩阵挖掘出有关特征基因。该算法避免了传统特征基因提取方法的单一性和片面性,从基因之间的关联性出发,使结果具有生物学意义。在此基础上,考虑到基因表达数据无法体现基因调控转录信息,所以将以上特征基因表达数据和转录因子调控靶基因信息数据进行整合,运用到NCA中得出了转录因子的表达活性及其对靶基因的调控强度,最终本文构建了在健康对照样本和AD患病样本中的转录调控网络。分子生物学分析表明,TF活性的变化及其对TG的调控影响在AD的发病和恶化过程里起着重要作用,可通过这些成果为探索AD的发病机制增加新的思路和依据。
1算法
1.1基于互信息的特征基因提取
对于复杂的基因关系,熵和互信息的方法能有效抓住基因与基因之间的关联性,能有效提取出复杂疾病的致病基因[6]。在信息论中,熵是用来衡量一个随机变量出现的期望值。设基因变量X=[x1,x2,…,xs]是一个基因表达模式,S代表基因表达数据中的样本的数量,则该基因表达模式的熵就是该模式所包含的信息量的度量。基因变量X的熵为:
(1)
联合熵是指一对基因变量X和Y的不确定的度量,即:
(2)
对于两个随机变量X和Y,其互信息I(X,Y)就是其中一个变量能提供给另一个变量的信息量,即:
(3)
根据上述公式得出互信息最终公式为:
I(X,Y)=H(X)+H(Y)-H(X,Y)
(4)
根据互信息值分析2个基因变量间的关联性,若互信息值较大,表明2个基因变量之间相关性较大,可认为这2条基因在生物学上存在着较强的关联性;反之,若互信息值为0或者较小,则表明这两条基因变量不相互包含任何信息,即在生物学上不存在关联性。对于基因变量间的互信息计算,本文借助直方图的思想[7],首先将基因表达数据全部离散化,分别求出基因的熵和基因之间的联合熵,再根据式(4)就能得出基因变量间的互信息。
说起帮衬二字,那当然是宝玉爹首先帮了宝刚爹的。没有宝玉爹的李代桃僵,就不会有宝刚爹一辈子的美满婚姻。真的,尽管在乡党们的眼里,宝刚爹是个怕老婆的标兵,在香娭毑面前,从没挺起胸抬起头做过人,可俗话说得好,鞋合不合脚,只有自己知道,宝刚爹对香娭毑,是打自心眼里敬畏和满意的。唯独不满意的一次,就是那次赛诗会,也就是香娭毑朗诵了爱毛主席的诗遭到宝刚爹的当场呛白之后,香娭毑赌气回南县老家侄儿那里住了好些日子,还是宝刚爹亲自去接才回到白家湾来。至于二狗伢说香娭毑与宝玉爹有那么一腿之事,乡党们似乎抱的是一种无所谓的态度,真也罢,假也罢,都有可能,都有道理,在乡下,是没有谁去认真追究的。
对于基因表达数据的微阵列矩阵E=(eij)N×S,其中N和S分别表示的基因的数量和数据样本数,eij代表的是第i条基因在第j个样本下的表达水平值。对于只包含两类状态的样本,可将E分为Econtrol和Eaffected两部分,即健康组和患病组样本下的基因表达数据。通过计算Econtrol和Eaffected中的每对基因间的互信息,最终分别得到形状为N×N的互信息矩阵Icontrol和Iaffected。基于互信息的性质,本文认为在健康组样本里和其它基因相关性较小,即互信息值较小的基因称为与其他基因失联,而在患病样本下该基因又与其他基因产生较大的相关性,即互信息值较高称之为与其他基因密切关联,则可以认为此类基因在疾病产生过程中从失联状态变化为表达异常且发挥着关联作用势必对疾病的产生具有重要作用,因此将此类基因作为特征基因。另外在疾病产生过程中,有些基因和其他基因的关系是从关联状态变化到失联状态,也将这些基因作为是特征基因,本文只考虑前者。提取此类特征基因的具体方法为:选择2个阈值Tcontrol和Taffected(Tcontrol>Taffected),并对Icontrol和Iaffected进行如下算法操作:
ifi==j,thenIcontrol(i,j)=0else ifIcontrol(i,j)≥Tcontrol,thenIcontrol(i,j)=1elseIcontrol(i,j)=0
(5)
ifi==j,thenIaffected(i,j)=0else ifIaffected(i,j)≤Taffected,thenIaffected(i,j)=1elseIaffected(i,j)=0
(6)
I(i,j)=Icontrol(i,j)&Iaffected(i,j)
(7)
式(5)和式(6)的目的是将互信息矩阵Icontrol和Iaffected进行二值化运算。为了剔除基因的自相关干扰,可以发现两式首先都对矩阵中的对角线元素进行了归0。为了更好地看出两类样本中基因之间的关联性变化,利用式(7)对两类互信息矩阵进行元素之间的逻辑“与”运算构建出了矩阵I,借助这个矩阵去挖掘特征基因。为了更形象化的分析,将矩阵I类比为图的邻接矩阵,显然每条基因就对应于图的一个顶点。若元素I(i,j)=1,即Icontrol(i,j)=1且Iaffected(i,j)=1也就是Icontrol(i,j)≥Tcontrol且Iaffected(i,j)≤Taffected,表明第i条基因和第j基因在健康对照样本中关联性较大,而在患病样本中关联性较小,即认为在图中表示这两点有线段连接。反之,若元素I(i,j)=0,则认为在图中表示这两点无线段连接,互相孤立。本文为了提取出在健康组样本中和其他基因关联度较小,而在患病样本中关联度较大的基因作为特征基因,很显然是挑出图中的这些孤立点。因为这些孤立点即基因,绝大部分是属于先在健康组样本中和其他基因关联度低甚至失联,而在患病样本关联度高的基因。通过对这些基因的研究,对探寻AD致病基因、信号传导通路及其转录调控过程具有重要意义。
1.2网络成分分析算法原理
由于提取的特征基因表达数据无法体现出基因调控转录信息,本文结合转录因子对靶基因调控的生物学先验知识,基于网络成分分析算法,构建基因转录调控网络。网络成分分析NCA是一种用来分析转录网络基因表达数据的算法,算法实质是根据基因表达数据和转录因子-靶基因调控关系的连通性网络,推导出转录因子活性TFAs(transcriptionfactoractivities)和转录因子对其靶基因的调控强度CS(controlstrengths)。转录调控模型如下:
(8)
式中Ei(t)代表基因表达水平,TFAi(t)(j=1,2,…,L)表示的是转录因子活性,CSij表示的是转录因子j对基因i的调控强度,(t)和(0)分别表示的是指定条件t和参考条件0。将对数-线性变换作为标准化方法来近似此非线性系统,通过对数转换后,式(8)的矩阵形式表示为:
[E]=[C][P]+Γ
(9)
式中矩阵[E](N×M)代表N个基因在M个样本下的基因表达矩阵,矩阵[C](N×L)表示的是转录因子对靶基因的调控强度矩阵以及矩阵[P](L×M)表示的是L个转录因子在M个样本下的表达活性矩阵,N表示基因数量,M表示实验样本个数以及L表示的是转录因子个数,此外Γ该模型的残差矩阵。若基因i不被转录因子j所调控,那么就将调控矩阵[C]中的元素Cij初始值设置为零;反之,则将元素Cij初始值设置为非零值。
由于基因表达矩阵[E]分解成因子矩阵并不具有唯一性,文献[4]已证明,若矩阵[C]和[P]满足唯一性条件,NCA算法能确保得到唯一解,来达到对任何给定的残差矩阵Γ的比例因子。此条件很明确地将NCA结果衔接到生物系统上,使解释简单明了。为了找出式(9)的最优解,最小二乘法约束被运用:
min‖[E]-[C][P]‖2s.tC∈Z0
(10)
式中矩阵Z0是连接模式矩阵,[C]和[P]的实际估计是通过两步交替最小二乘算法实现,该算法利用的是线性分解的双凸性。最小二乘约束法等价于具有独立同分布成分的高斯噪声存在下的最大似然方法。对于NCA算法,详见文献[4]。
2仿真结果与分析
2.1基于互信息算法的特征提取结果分析
本文选用的实验数据是来自美国国立生物技术信息中心NCBI(nationalcenterforbiotechnologyinformation)网站的基因表达综合数据库GEO(geneexpressionomnibus,http://www.ncbi.nlm.nih.gov/gds/)中的数据集GSE5281。该AD数据集是由LiangWS等人提供的,包含了161组不同大脑皮层组织样本的基因表达数据[8],本文选用的是海马区HIP(Hippocampus)基因表达数据,它拥有13个健康对照样本和10个患病样本及每组样本包括54 675个基因探针所对应基因表达数据。
对于数据的预处理,本文首先将数据进行归一化,目的是将所有基因表达数据值都映射到[-1,1]区间中,然后通过公式(1)计算每条基因的信息熵,挑出信息熵值大的基因。将信息熵应用于基因表达数据,如某基因信息熵越大,表明该基因在样本中含有的信息量越大,分类贡献率也就越大;反之,信息熵越小,该基因信息量越小,分类贡献率越低。最终通过计算,选择了5000条基因。分别计算这5000条基因在健康对照样本和患病样本中的互信息值,得到了两个互信息矩阵,既Icontrol和Iaffected。对于阈值Tcontrol和Taffected的选取是本文基于互信息方法提取特征基因的关键。若Tcontrol值选取过大同时Taffected值选取过小,会造成得到的特征基因数量过多;若Tcontrol值选取过小同时Taffected值选取过大,会造成得到的特征基因数量过少,甚至提取不到AD易感基因。这些问题都将直接影响本文后面基于NCA算法调控网络的构建。对得到的这两个互信息矩阵进行式(5)-式(7)处理,经过多次验证,最终设定Tcontrol=2.3,Taffected=1.7得到实对称矩阵I,通过对该邻接矩阵的每行或每列进行求和运算。和值越大,表明这条基因在健康组样本中和许多基因关联,反之和值为0的基因,则说明它们在健康组样本中并不和其它基因关联,且它们绝大部分是在患病样本中和许多基因关联的。通过剔除和值非0的点,本文挖掘出了493个基因作为特征基因。这些基因中绝大部分是在健康对照样本里与其它基因失联,而在AD患病样本中却和其他基因强关联。
2.2NCA结果分析
通过NCA算法推断转录因子的活性和转录因子对靶基因的调控强度,目的是为了挖掘出在健康对照和AD患病样本中的转录调控网络。本文选用网站BIOBASE(http://www.gene-regulation.com)中的TRANSFAC公共数据库,该数据库拥有许多现有的转录因子调控靶基因的信息。为了找寻AD有关的关键转录因子,本文将前面互信息提取方法所得到的重要特征基因和该TF-TG调控关系数据库进行匹配,选择了调控特征基因数量最多的前17个转录因子。表1给出了这17个转录因子及其所对应调控的靶基因。对健康对照和AD患病样本数据,分别运行NCA之前,需要建立两个输入:一个是矩阵[E],它表示的是原始AD基因表达数据所提供的在健康对照或患病样本中的靶基因基因表达谱;另一个是预定义的初始连接矩阵[C0],它表示的是转录因子对靶基因的调控关系,若TF调控TG,则令其对应的连接矩阵[C0]中的元素值为1,反之值为0。通过NCA算法,最终筛选出了17个转录因子和46个靶基因,并且由此得出了转录因子分别在健康组样本和患病样本下的表达活性和其对靶基因的调控强度。
基因的转录是通过一小部分转录因子控制的,通过翻译后修饰或配体结合过程,它们的表达活性对于基因的表达水平来说是决定性的因素。一般而言,转录因子活性水平(TFAs)并不总是和它们的基因表达谱TFEV(geneexpressionvaluesofTF)呈现出正相关。图1给出了所有转录因子的表达活性和它们在AD原始微阵列数据中的基因表达谱的变化对比,其中端点带有圆形标注的线段表示的是转录因子在不同样本中的表达活性变化;而带有上三角形标注的线段表示的是转录因子在不同样本中的基因表达水平变化。另外图1,X轴中点“1”对应健康对照样本,点“2”对应患病样本及Y轴对应转录因子活性或基因表达水平值。图1每个子图都显示了转录因子在不同病程样本里的活性和基因表达强度,可以明显发现从健康样本到患病样本过程中,表达活性明显下降的转录因子有:ANAPC5,BUB3,DRAP1,MCM4,NAT13,THOC4,ZNF317;表达活性明显上升的转录因子有:G3BP1,HNRPD,MRPL44,MRPS12,NLRP1,RFC5,ZBTB20。另外从转录因子的活性和基因表达强度的相关性分析,呈现正相关性的有:ANAPC5,BUB3,DRAP1,G3BP1,MCM4,MRPL44,MRPS12,NAT13,NLRP1,ZNF317,THOC4,ZBTB20;呈现负相关性的有:HNRPD,RFC5。

表1 选取的转录因子与其对应的靶基因
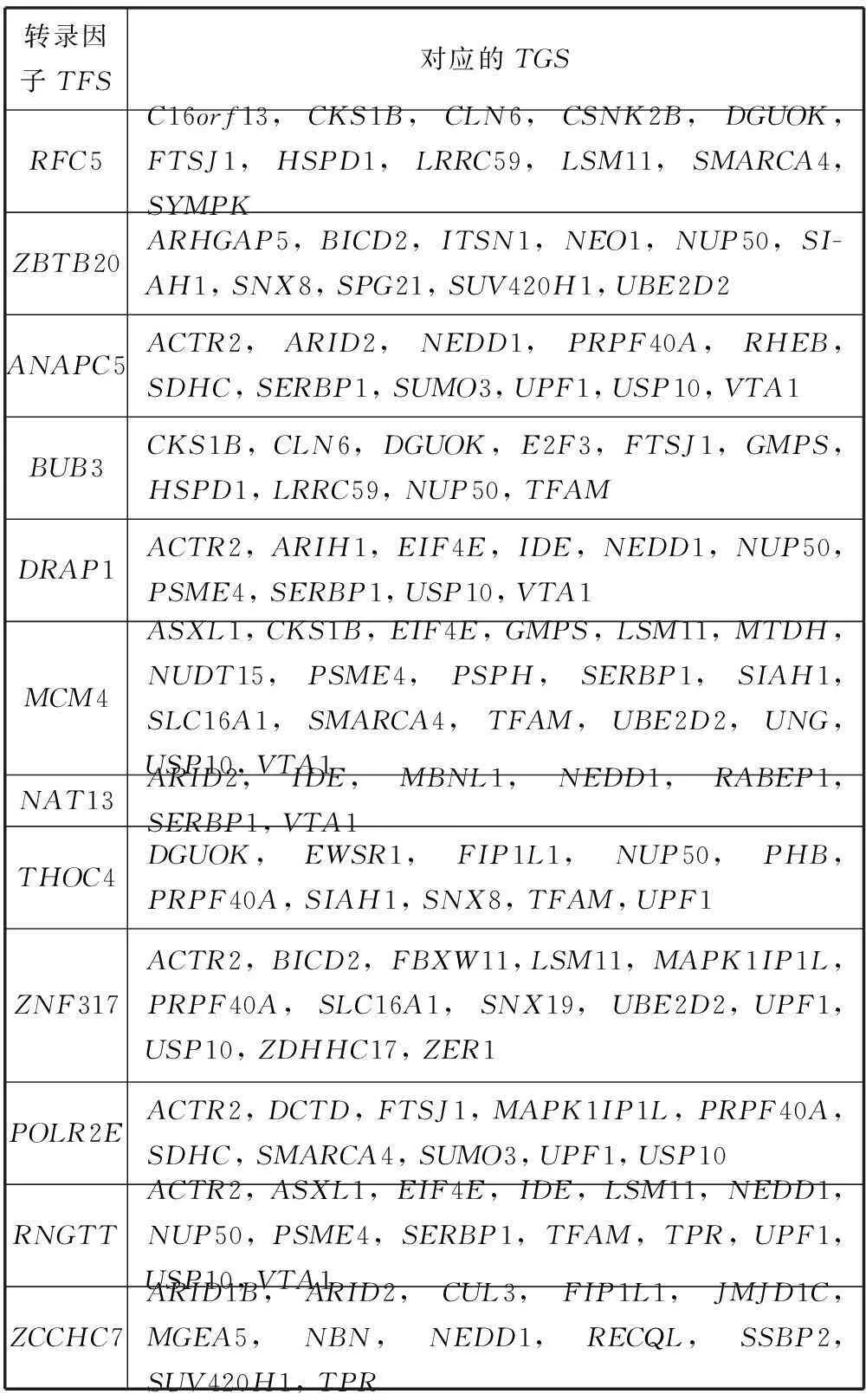
续表1
2.3AD动态调控网络分析
基于NCA算法所得到的转录因子活性矩阵[P],调控矩阵[C]以及原始微阵列靶基因表达数据,本文构建了在健康对照样本和AD患病样本下的转录调控网络图,如图2和图3所示。为了更加形象地看出所有转录因子及靶基因的动态变化趋势,对构建调控网络所用到的数据首先都进行了归一化,其中靶基因的表达值由矩阵[E]中基因在对应样本中取均值所得,转录因子的表达活性由矩阵[P]中转录因子在对应样本中取均值所得,而转录因子对靶基因的正负调控作用由矩阵[C]中转录因子对靶基因的调控值的正负来定性表示。图2和图3中圆形节点表示转录因子,方形节点表示靶基因,三角形节点表示AD易感基因;节点颜色越深表示其表达水平越高或越低;连线则表示转录因子对靶基因有正负调控作用。
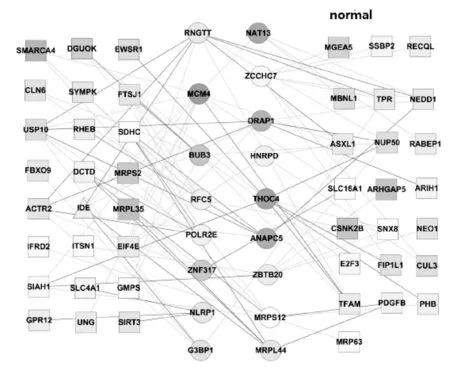
图2 健康对照样本转录调控网络图

图3 AD患病样本转录调控网络图
从构建的转录调控网络图以及表1可见,多个转录因子可以共同调控一个靶基因,多个靶基因也可以被一个转录因子共同调控,即靶基因的表达受到一个或多个转录因子的表达活性的综合影响。如图中转录因子DRAP1,G3BP1,MRPL44,NAT13,RNGTT共同调控靶基因IDE,与健康组样本对照,可以看出IDE的表达水平下降(颜色从深色到浅色),而转录因子G3BP1在患病样本中被极大的激活(颜色深度加重),其中IDE是目前已知的AD易感基因[9],G3BP1对肺癌和乳腺癌扩散具有中介效应[10,11],并且文献[11]指出RAS-GAPSH3结构域结合蛋白(G3BP)是基因USP10的调制器,巧合的是网络中G3BP1调控的多个靶基因中就包括基因USP10,与健康组样本对照,可以看出USP10的表达水平升高。从中可以推断,G3BP1与AD的发生密切相关。转录调控图中如靶基因TFAM被转录因子THOC4,BUB3,MCM4,RNGTT共同调控,与健康组样本对照,可以看出TFAM的表达水平升高(颜色从深色到浅色),BUB3在AD患病样本中活性明显降低;其中线粒体转录因子(TFAM)多态性与AD有关[12],BUB3是构成有丝分裂纺锤体配置复合物的关键组成部分,能生成其他重要蛋白复合物[13]。在健康对照和AD患病网络图中特定的转录因子对于同一个靶基因的调控影响作用不一定相同,如转录因子NLRP1对于靶基因GPR12的调控一直都是促进作用;然而转录因子ANAPC5对于靶基因ACTR2的调控作用,在健康组样本中对其是抑制的,而在患病样本中对其表达起促进作用。其中基因GPR12涉及到细胞增殖和存活的调控[14],ACTR2又名ARP2,文献[15]指出ARP2/3的丢失会导致趋化信号传导中的NF-κB依赖性,是非自治的影响。炎性体(Inflammasomes)是专门的信令平台,对于先天免疫和炎症反应的调节至关重要,各种NLR家族成员(如NLRP1,NLRP3和IPAF等)以及PYHIN家族成员AIM2可形成炎性体复合物。ChoiAJ等人发现了激活NLRP3炎性信号通路的调控机制,并讨论了在代谢和认知性疾病,包括肥胖症、2型糖尿病、阿尔茨海默症(AD)和抑郁症中NLRP3的潜在作用[16]。对于转录因子ANAPC5,与健康组样本对照发现,其在患病样本中活性被抑制,它是APC的一个亚基,并且APC对于细胞在分裂后期退出有丝分裂过程以及防止其过早进入DNA合成期(S期)有着重要作用[17]。通过在线基因分析网站DAVID(http://david.abcc.ncifcrf.gov/)对涉及到的所有靶基因和转录因子进行定性分析,该网站包括许多基因分析数据库,如常见的KEGG[18](KyotoEncyclopediaofGenesandGenomes)和GO[19](geneontology)数据库。针对KEGG通路发现,ANAPC5不仅和BUB3,MCM4,E2F3形成了细胞周期通路,也和其它基因参与了泛素介导的蛋白水解过程通路,这也进一步证明了ANAPC5对于AD的发病机理可能起着重要作用。
此外,对于转录调控网络的定性分析,也发现了许多与常见癌症密切相关的靶基因和转录因子。如转录因子HNRPD又名AUF1,它的表达与肝癌的恶化过程有着密切关系、对于在淋巴结阳性乳腺癌患者中EIF4E的高表达可能是全身扩散的标志和抑制素(PHB)表现为细胞增殖的负调节剂以及是一种肿瘤抑制剂以及SIRT3是作为乳腺癌肿瘤抑制蛋白等。针对以上这些分析结果可知,AD与癌症或者其它疾病之间其实并不是彼此孤立的,它们的致病机理可能是有内在联系的。本文通过对AD调控网络的全面生物学分析,发现了一些AD致病原因,其中发现它们中有一些都与有丝分裂、细胞生长、免疫反应和炎症反应有着密切关系。在以后的研究里,将集中围绕与AD有关的有丝分裂、细胞生长、免疫反应和炎症反应过程,为的是去发现真正的AD致病机理。
3结语
微阵列高通量技术的运用,使得所有mRNA转录产物可以同时测量,从而让构建基因调控网络成为可能。传统的基因网络分析模型如ICA并不能运用转录调控信息,并且只是纯粹的基于调控信号的数学和统计特性,因此它们并不能准确地构建调控网络。本文从基因之间的关联性出发,首先利用互信息(MI)提取特征基因,探寻AD发病过程中明显发生关联作用的基因组;然后将所得的特征基因与转录调控信息进行融合挖掘出AD关键转录因子;最后通过NCA算法推断出转录因子在不同样本下的活性和其对靶基因的调控强度,并成功构建出转录调控网络。通过对AD调控网络的分子生物学分析,发现了与AD密切相关的基因IDE和TFAM;与细胞周期有关的基因ANAPC5,BUB3 等;与炎症反应有关的基因NLRP1等。这些基因以及生物过程都与AD的致病机理有着紧密的联系。另外,通过AD调控网络分析,本文也发现了许多与癌症有关的基因。这些发现可能有助于为AD致病机理的研究提供新的依据和方法。
参考文献
[1]BrookmeyerR,JohnsonE,Ziegler-GrahamK,etal.ForecastingtheglobalburdenofAlzheimer’sdisease[J].Alzheimer’s&dementia,2007,3(3):186-191.
[2]KaissiO,NimpayeE,SinghTR,etal.GenesSelectionComparativeStudyinMicroarrayDataAnalysis[J].Bioinformation,2013,9(20):1019.
[3]HyvärinenA,OjaE.Independentcomponentanalysis:algorithmsandapplications[J].Neuralnetworks,2000,13(4):411-430.
[4]LiaoJC,BoscoloR,YangYL,etal.Networkcomponentanalysis:reconstructionofregulatorysignalsinbiologicalsystems[J].ProceedingsoftheNationalAcademyofSciences,2003,100(26):15522-15527.
[5] 张焕萍,王惠南,卢光明,等.基于互信息的差异共表达致病基因挖掘方法[J].东南大学学报:自然科学版,2009,39(1):151-155.
[6] 孙啸,陆祖宏,谢建明.生物信息学基础[M].清华大学出版社有限公司,2005.
[7]SteuerR,KurthsJ,DaubCO,etal.Themutualinformation:detectingandevaluatingdependenciesbetweenvariables[J].Bioinformatics,2002,18(suppl2):S231-S240.
[8]LiangWS,ReimanEM,VallaJ,etal.Alzheimer’sdiseaseisassociatedwithreducedexpressionofenergymetabolismgenesinposteriorcingulateneurons[J].ProceedingsoftheNationalAcademyofSciences,2008,105(11):4441-4446.
[9]CuiPJ,CaoL,WangY,etal.Theassociationbetweentwosinglenucleotidepolymorphismswithintheinsulin-degradingenzymegeneandAlzheimer’sdiseaseinaChineseHanpopulation[J].JournalofClinicalNeuroscience,2012,19(5):745-749.
[10]WinslowS,LeanderssonK,LarssonC.RegulationofPMP22mRNAbyG3BP1affectscellproliferationinbreastcancercells[J].Molecularcancer,2013,12(1):156.
[11]SonciniC,BerdoI,DraettaG.Ras-GAPSH3domainbindingprotein(G3BP)isamodulatorofUSP10,anovelhumanubiquitinspecificprotease[J].Oncogene,2001,20(29):3869-3879.
[12]ZhangQ,YuJT,WangP,etal.MitochondrialtranscriptionfactorA(TFAM)polymorphismsandriskoflate-onsetAlzheimer’sdiseaseinHanChinese[J].Brainresearch,2011,1368:355-360.
[13]KumarA,RajendranV,SethumadhavanR,etal.CEPproteins:theknightsofcentrosomedynasty[J].Protoplasma,2013,250(5):965-983.
[14]LuX,ZhangN,MengB,etal.InvolvementofGPR12intheregulationofcellproliferationandsurvival[J].Molecularandcellularbiochemistry,2012,366(1-2):101-110.
[15]WuC,HaynesEM,AsokanSB,etal.LossofArp2/3inducesanNF-κB-dependent,nonautonomouseffectonchemotacticsignaling[J].TheJournalofcellbiology,2013,203(6):907-916.
[16]ChoiJS,RyterSW.Inflammasomes:MolecularRegulationandImplicationsforMetabolicandCognitiveDiseases[J].Moleculesandcells,2014,37(6):441-448.
[17]LatchmanDS.Transcriptionfactors:anoverview[J].Internationaljournalofexperimentalpathology,1993,74(5):417.
[18]KanehisaM,GotoS,KawashimaS,etal.TheKEGGresourcefordecipheringthegenome[J].Nucleicacidsresearch,2004,32(suppl1):D277-D280.
[19]YangAC,HsuHH,LuMD.Applyinggeneontologytomicroarraygeneexpressiondataanalysis[C]//SystemScienceandEngineering(ICSSE),2010InternationalConferenceon.IEEE,2010:421-426.
SIGNIFICANT GENES EXTRACTION BASED ON MUTUAL INFORMATION ANDTRANSCRIPTIONALREGULATORYNETWORKRECONSTRUCTION
Kong Wei1Zhi Xing1Mou Xiaoyang2
1(Information Engineering College,Shanghai Maritime University,Shanghai 201306,China)2(Department of Chemistry and Biochemistry,Rowan University,NJ 08028,USA)
AbstractTraditional feature genes extraction methods tend to focus only on the expression difference of a single gene in different samples, but ignore the correlation among genes as well as the links between multiple pathogenic genes as one gene module and complex diseases. In view of this, we proposed a mutual information-based feature genes extraction algorithm, it is used to extract those genes that have the most significant differences and changes in correlation between the healthy controls and Alzheimer’s disease (AD) samples. On this basis, in combination with the biological priori information about the regulatory of transcription factors (TF) on target gene (TG), we applied network component analysis algorithm (NCA) in analysing TF’s expression activities and their regulatory strengths on TGs, and constructed the transcriptional regulatory networks of AD feature genes. Molecular biology analysis showed that the changes of them in mitosis, cell cycle, immune response and inflammation play an important role in deterioration of AD.
KeywordsAlzheimer’s disease (AD)Mutual information (MI)Transcription factor (TF)Network component analysis (NCA)
收稿日期:2014-12-31。国家自然科学基金项目(61271446)。孔薇,教授,主研领域:生物信息处理,模式识别。支星,硕士生。牟晓阳,教授。
中图分类号TP391.9Q343.1
文献标识码A
DOI:10.3969/j.issn.1000-386x.2016.06.057
