新型Ⅰ型普鲁兰酶基因的克隆表达及酶学性质*
2016-07-18王青艳申乃坤朱绮霞谢能中黄日波
王青艳,申乃坤,朱 婧,秦 艳,朱绮霞,谢能中,李 亿,黄日波
(广西科学院,国家非粮生物质能源工程技术研究中心,非粮生物质酶解国家重点实验室,广西生物炼制重点实验室,广西南宁 530007)
新型Ⅰ型普鲁兰酶基因的克隆表达及酶学性质*
王青艳,申乃坤,朱婧,秦艳,朱绮霞,谢能中,李亿,黄日波**
(广西科学院,国家非粮生物质能源工程技术研究中心,非粮生物质酶解国家重点实验室,广西生物炼制重点实验室,广西南宁530007)
摘要:【目的】筛选并克隆表达高酶活且具有一定热稳定性的新型普鲁兰酶。【方法】克隆Tumebacillus flagellatus GST4的普鲁兰酶基因pulB,构建重组质粒后转化宿主菌大肠杆菌进行诱导表达,再运用亲和层析进行纯化并分析其酶学性质和结构。【结果】pulB在大肠杆菌中实现可溶性表达,发酵液上清酶活力达到78 U/mL,粗酶液经纯化后比活力为258 U/mg。重组酶PulB最适反应温度和pH值分别为55℃和5.0,在较窄的酸性范围内(pH值4.5~5.5)酶活力比较稳定;对普鲁兰糖的Km=(16.28±0.03)mg/mL,Vmax=(22.05±0.02)μmol·min-1·mg-1。PulB的DNA序列与GenBank数据库里的任何序列都没有同源性,在蛋白质序列上,由基因pulB编码的氨基酸序列与T.aegyptius的环麦芽糖糊精酶相似性最高,BlastX比对的Identities为54%,Positives为69%,SMART结构预测分析发现,pulB具有淀粉酶的结构域。底物特异性分析表明,它可水解普鲁兰糖和支链淀粉生成线性的低聚糖或麦芽三糖。【结论】重组酶PulB是尚未报道的新型普鲁兰酶,它可水解普鲁兰糖和支链淀粉,属Ⅰ型普鲁兰酶。
关键词:Ⅰ型普鲁兰酶膨胀芽孢杆菌克隆表达酶学性质
0引言
【研究意义】普鲁兰酶(Pullulanase,EC 3.2.1.41)是α-淀粉酶家族GH13中的一种脱支酶,它能够专一性的水解普鲁兰、淀粉和糖原中的α-1,6-糖苷键[1]。自然界中的淀粉大多为支链淀粉[2],其中α-1,6-糖苷键占4%~5%。普鲁兰酶能分解支链的特性决定了它在淀粉加工业中的广泛应用,比如:在淀粉水解过程中专一性切开支链淀粉分支点中的α-1,6-糖苷键,从而剪下整个侧枝形成直链淀粉,后者具有很好的抗水性和成膜性,有望用于生产可食性包装膜,解决“白色污染”问题;Ⅰ型普鲁兰酶能够与α-淀粉酶、β-淀粉酶等协同作用生产高葡萄糖浆、超高麦芽糖浆、提高啤酒中的发酵度;普鲁兰酶和α-淀粉酶以及糖化酶协同作用,可在以非粮木薯为原料生产酒精的液化和糖化过程发挥作用,普鲁兰酶要求的底物分子结构最小,它可以将最小单位的支链分解,最大限度地利用淀粉原料,加速糖化过程,从而有效的提高淀粉利用率和水解效率[3]。除此之外,采用普鲁兰酶将各类淀粉脱支后再糊化老化处理可增加抗消化淀粉的含量,后者对人体血糖水平、肥胖、癌症、脂质代谢和能量等方面有重要生理功能[4]。而目前工业应用上的挑战是能否找到满足低成本高产、且酶学性质(如耐酸、耐热)符合工业应用要求的普鲁兰酶。因此,通过筛选高产菌株、克隆异源表达以及突变普鲁兰酶基因等方式,获得所需的普鲁兰酶,具有重要的应用研究意义。【前人研究进展】普鲁兰酶最初是由Bender和Wallenefls于1966年通过产气杆菌(Aeorbaetere aerogenes)发酵获得[5]。此后,各国的科研人员经过广泛深入研究,从不同的地区微生物中获得该酶。1975年日本的Yoshiyuki Takaskai[6]发现蜡状芽抱杆菌覃状变种(Bacillus cereus Var.mycodes)能产普鲁兰酶,该酶的最佳作用条件为pH值6.0~6.5,温度50℃。20世纪80年代初,丹麦Novo公司以嗜酸性分解普鲁兰多糖芽孢杆菌(Bacillus acidopullulyticus)[7]研发出耐热(60℃)耐酸(pH值4.5)的商品普鲁兰酶Pormozyme,是目前应用最广且产量最大的普鲁兰酶。20世纪90年代,Deweer等[8]发现普鲁兰酶产生菌Bacillus naganoensis;Tomimura等[9]筛选出Bacillus deramificans,这两株菌与B.acidopullulyticus所产普鲁兰酶的酶学性质相似,其发现进一步拓宽了普鲁兰酶的应用。随着基因工程技术的发展,利用其构建基因工程菌,以提高目的蛋白的产量并减少下游工艺的成本,这在世界范围内越来越广泛地得到重视并加以应用。1984年,日本科学家在大肠杆菌中成功表达Klebsiella aerogenes普鲁兰酶基因,但酶活力水平不高且在非选择性培养基中表现型极不稳定。1985年,Takiazwa把普鲁兰酶基因(包括结构基因和操纵基因)克隆入多拷贝载体pBR322,得到比野生菌株酶活力水平高20~40倍的工程菌,此工程菌能保持高酶活力水平两周。上述这些酶大部分都是不能分泌到胞外的胞内酶。1999年,Tomimura从Bacillus naganoensis (ATCC53909)中分离出普鲁兰酶基因,并在原核生物表达系统枯草芽抱杆菌中成功表达,所产生的重组普鲁兰酶具有很好的应用特性[10]。该酶在60℃时测得最适反应pH值为5.0,在pH值为4.5条件下测得最适反应温度为60℃;在pH值4.5,60℃保温55 h仍有50%的残余酶活力,具有较好的热稳定性。此后,特别是进入20世纪90年代后,相继有许多耐热普鲁兰酶基因被克隆、测序并在大肠杆菌和枯草杆菌中表达,有的还申请专利保护[10-11]。目前已有数十个普鲁兰酶基因在以大肠杆菌为主的表达系统中实现异源表达。通过基因工程手段表达并提高普鲁兰酶产量是普鲁兰酶基础研究和应用研究的主要发展方向。另外,淀粉加工时通常采取55~60℃的反应温度,这就对普鲁兰酶在该温度下的物理化学稳定性提出较高的要求。【本研究切入点】虽然基因工程技术使得普鲁兰酶的异源表达研究取得了一些成绩,但总体来说表达水平不高。我国从20世纪70年代开始便研究开发普鲁兰酶,但是到目前仍然仅限于实验室研究并且酶活力较低[12-13],一般为2~5 U/mL,这些酶的产生菌株大多为病原菌,其发酵液不能直接用于工业添加,特别是食品工业,因此普鲁兰酶基因的重组表达至关重要。【拟解决的关键问题】本研究采用Novagen公司的pET表达系统异源表达T.flagellates GST4普鲁兰酶基因,并进行酶学性质和结构预测分析。通过异源可溶性表达提高酶的产量,获得新的Ⅰ型普鲁兰酶并初步探讨其结构与功能的关系。
1材料与方法
1.1菌株和质粒
菌株T.flagellates GST4为本实验室从位于南宁的木薯淀粉废水中筛选所得[14],大肠杆菌E.coli DH5α(本实验室保存)用于常规的克隆和质粒繁殖宿主,大肠杆菌E.coli BL21(DE3)(Stratagene)为表达宿主。pET-22b(+)(Novagen)是具有氨苄抗性、N端信号肽pelB和C端6-His 标签的表达载体,pMD-18T(TaKaRa)为T-载体。
1.2试剂
Prime START聚合酶、rTaq DNA聚合酶、各种限制性内切酶、dNTPs、T4 DNA连接酶、小牛肠性磷酸酶(CIP)、DNA连接试剂盒、DNA Maker DL2000、λ/HindⅢ DNA Marker和蛋白质分子量标记均购自TaKaRa公司,蛋白酶K、RNA酶A和DMSO 购自Fermentas公司,细菌基因组DNA提取试剂盒、质粒小量提试剂盒和凝胶回收试剂购自北京天根公司,镍填充料Chelating Sepharose购自Amersham Biosciences公司,Sephadex G200购自Pharmacia公司,Bradford蛋白质检测试剂盒购自Generay,蛋白质电泳预制胶购自Bio-rad,咪唑购自上海生工生物工程有限公司,普鲁兰糖、可溶性淀粉和支链淀粉等购自国药集团化学试剂有限公司,引物合成和测序均由上海生物工程公司完成。
1.3培养条件
挑取单菌落于LB液体培养基中,37℃,220 r/min摇床振荡培养过夜活化,用接种环在固体平板划线,固体平板倒置于37℃恒温箱培养过夜。挑取单菌落于新鲜的LB液体培养基中摇床培养约12 h后,按1%(V/V)的接种量接种于新鲜的LB液体培养基中,37℃,220 r/min培养约3~4 h后,加入IPTG使其终浓度为0.5 mmol/L,继续培养12~16 h,收集菌液。
1.4总DNA和质粒的提取
新鲜平板上挑取单菌落接入液体培养基,过夜培养,收集菌体细胞用于总DNA和质粒DNA提取,提取方法参照文献[15]。
1.5表达载体的构建
1.5.1pulB的克隆和基因序列分析
根据已经测序T.flagellatus GST4基因组的注释为假设普鲁兰酶的基因序列设计引物,所用的上下游引物(下划线分别为引入的酶切位点BamHⅠ和HindⅢ)序列为
pulB-F:5′-CGCGGATCCCAATCAGGAAG-CTATTTTTCA-3′;
pulB-R:5′-ACCAAGCTTCACCGTTCCGCCGCTCA-3′。
以T.flagellatus GST4的总DNA为模板,进行PCR扩增,PCR反应体系为50 μL反应液中含有5×PS PCR缓冲液10 μL,dNTPs 200 μmol/L,总DNA模板10 ng,引物各3.2 pmol,PrimeSTAR DNA聚合酶5 U。扩增程序为95℃预变性2 min;98℃ 10 s,53.6℃ 15 s,72℃ 2 min,共30个循环延伸;72℃,10 min。
扩增成功后用OMEGA公司Extraction Kit 纯化普鲁兰酶基因,pulB回收的片段一部分经TA克隆之后与T载体pMD18-T连接,获得pMD-pulB用于测序;另一部分作下一步的双酶切。
基因pulB的序列用megablast (highly similar sequences) 软件检索GenBank核苷酸数据库[16];用BlastX软件检索GenBank氨基酸数据库;用在线SMART(Simple Modular Architecture Research Tool,http://smart.embl.heidelberg.de)工具分析预测该基因编码蛋白质序列的组件结构和功能域[17]。
1.5.2pulB与载体的酶切、连接和转化
DNA片段与载体的酶切、连接和转化参照文献[15],将PCR扩增得到的pulB目的片段用BamHⅠ/HindⅢ双酶切,与经同样双酶切的质粒载体pET-22b(+)的大片段混合,用连接酶于16℃连接过夜,连接产物转化宿主大肠杆菌感受态细胞E.coli DH5α,冰上放置30 min,42℃水浴热击90 s,加入600 μL经42℃预热的SOC培养基,于37℃,200 r/min预培养30 min,取适量涂布于含氨苄抗性的LB平板,37℃恒温箱倒置培养过夜。转化子提取质粒后用BamHⅠ/HindⅢ进行双酶切验证,同时以转化子质粒为模板,用原扩增引物进行PCR验证。
1.6重组质粒pET-pulB的诱导表达和纯化
1.6.1诱导表达
将重组质粒pET-pulB转化表达宿主E.coli BL21(DE3)感受态细胞,挑取转化子用含合适氨苄的LB液体培养基过夜培养12 h,按1%(V/V)的接种量转接到50 mL的LB(含100 μg/mL氨苄)培养基中,37℃,220 r/min摇床振荡培养至OD600为0.4~0.6时加入IPTG(终浓度为0.5 mmol/L),在25℃,220 r/min继续培养14 h,离心收集上清,上清装入透析袋,外敷PEG6000置于4℃冰箱中进行透析浓缩,至透析袋中的液体量剩余约为10 mL时转入干净的50 mL离心管,4℃,8 500 r/min离心10 min,所得上清液即为粗酶液。
1.6.2重组酶的纯化
通过重组酶C端的6-His标签与镍离子的螯合进行纯化[18],纯化过程尽量在较低温(4~10℃)下进行。金属鳌合物的亲和层析-镍(IMAC)柱含5 mL 柱床体积的螯合琼脂糖凝胶(FF)介质,介质经NiCl2装载平衡后用超纯水洗涤,用25 mL平衡缓冲液(300 mmol/L NaCl,5~10 mmol/L咪唑,50 mmol/L pH值为7.4磷酸缓冲液)平衡柱子,粗酶液调节至与平衡缓冲液相同的pH值后上柱,再依次用25 mL咪唑浓度分别为20 mmol/L和60 mmol/L的平衡缓冲液洗涤杂蛋白,目的蛋白用25 mL 咪唑浓度为250 mmol/L 的平衡缓冲液洗脱,收集含目的蛋白质的洗脱液,用截留分子量为30 000 Da的Millipore超滤离心管进行缓冲液置换,所得的纯酶溶解在pH值为5.0的50~100 mmol/L磷酸钠-柠檬酸缓冲液中,并加入终浓度为10%(V/V)的甘油做保护剂,用于进行SDS-PAGE分析、酶活力测定和酶学性质分析。
1.7蛋白质含量检测
蛋白质含量测定参照Bradford法[19]进行,采用BRADFORD蛋白浓度测定试剂盒测定样品蛋白浓度,以牛血清蛋白(BSA)为蛋白质标准,测定OD595处的吸光值。具体方法参照试剂盒说明书。
1.8SDS-PAGE蛋白质电泳
取20 μL菌液/酶液于2 mL EP管,加入等体积的上样缓冲液,100℃水浴10 min,12 000 r/min离心5 min,取上清进行SDS-PAGE电泳,蛋白质电泳采用金斯瑞ExpressPlusTM预制胶(分离胶浓度10%(V/V),浓缩胶5%(V/V),电泳仪/槽使用Bio-Rad-PROTEAN(II/3/Tetra System)Hoefer Mighty Small(SE250/SE),电泳条件:电压120 V,起始电流75~100 mA,结束电流30~50 mA,时间1.0 h,电泳结束后用考马斯亮蓝染色,洗脱后观察。
1.9重组酶活力测定和酶学性质分析
1.9.1酶活力测定
采用3,5-二硝基水杨酸法(DNS法),反应体系及方法:取适当稀释的酶液100 μL,添加至400 μL醋酸-醋酸钠缓冲液(pH值为4.8,含质量体积比为1%的普鲁兰糖)中,55℃条件下反应10 min,加入500 μL DNS终止反应后于沸水浴煮沸5 min,冷却,用酶标仪在540 nm波长下检测反应液的吸光度值。
酶活单位定义:在相应条件,每分钟分解普鲁兰糖所释放的还原碳水化合物,其还原力即相当于1 μmol葡萄糖所需的酶量,用1 U表示。
酶活力计算公式:酶活力(U/mL)=
N(OD540+0.108)/(6.619×180×10-3)。其中N表示酶液稀释倍数;180表示葡萄糖分子量。
普鲁兰酶比活力计算公式:普鲁兰酶的比活力(U/mg)=普鲁兰酶活力/普鲁兰酶蛋白含量。
1.9.2最适反应温度和温度稳定性的测定
以1%(W/V)普鲁兰糖作为底物,在pH值为6.0条件下,于不同的温度(35~70℃,梯度为5℃)条件下测定温度对重组酶活力影响,并以最高酶活力计算各个温度下的相对酶活力,相对酶活力最高的温度就是该酶的最适作用温度。
在pH值为6.0条件下,将酶分别于37℃,42℃,47℃,58℃,62℃,68℃保温1 h后测定酶活力,与未经处理的酶活力的比值绘制温度稳定性曲线。
1.9.3最适反应pH值和pH稳定性的测定
以1%(W/V)普鲁兰糖作为底物,在最适反应温度、pH值为3.6~6.0(每隔0.2个单位)的磷酸钠-柠檬缓冲液(50 mmol/L)中测定重组酶的活力,并以最高酶活力计算各pH值下的相对酶活力,相对酶活力最高的pH值即为该酶的最适作用pH值。
将酶在pH值为3.5~7.0(每隔0.5个单位)的缓冲液里置37℃水浴条件保温24 h后测定重组酶的酶活力,与未经处理的酶活力的比值绘制pH稳定性曲线。
1.9.4Km和Vmax的测定
以不同浓度的普鲁兰糖为底物,在最适的反应温度和pH值条件下,测定普鲁兰酶PulB的酶活力,参考Malle等[20]的方法,利用Lineweaver-Burk作图法绘制1/V与1/[S]的双曲线图,并通过非线性回归方程计算得出重组酶的Km和Vmax的动力学参数。
1.9.5金属离子对酶活力的影响
在最适pH值和最适温度条件下,PulB在含有1 mmol/L的不同金属离子(Ba2+、Ca2+、Co2+、Cu2+、Mg2+、Zn2+、Pb2+、Mn2+、Fe2+)的1%(W/V)的普鲁兰糖溶液中作用30 min后,采用DNS法测定PulB酶活力,以不加金属离子的酶液作为对照,从而确定各种金属离子对普鲁兰酶活力的影响。
1.9.6底物特异性分析
用50 mmol/L,pH值为5.0的柠檬酸-磷酸钠缓冲液配制1%(W/V)的普鲁兰糖、支链淀粉、α-环糊精、直链淀粉和可溶性淀粉作为底物溶液,在最适反应条件下反应24 h,然后将反应产物100℃水浴10 min灭酶活,进行酶活力和HPLC酶水解产物分析。HPLC测试条件:戴安Utimat3000,自动进样器,色谱柱为AminexHPX-87H 300 mm×7.8 mm(有机酸柱),流动相:5 mmol/L H2SO4,pH值为2.5,柱温45℃,进样量10 μL,流速0.6 mL/min,示差检测器(Shodex),检测器温度为45℃。
2结果与分析
2.1普鲁兰酶基因pulB的克隆
以T.flagellatus GST4的总DNA为模板,用pulB-F和pulB-R引物特异地扩增出一条大约为1.7 kb的DNA条带,大小与预期的pulB基因的编码区相符(图1)。
2.2pulB基因序列分析和蛋白质结构预测
重组质粒pMD-pulB的普鲁兰酶基因测序结果表明,其开放读码框由1 728个核苷酸组成(附件1),G+C百分含量为56.4%,可编码由575个氨基酸组成的蛋白质。该蛋白质的预计分子量为66 672 Da,等电点pI为5.25。该基因序列经megablast(highly similar sequences) 软件检索GenBank核苷酸数据库没有发现任何同源序列,说明pulB基因是一个尚未报道过的基因。

M:DL2000 DNA 分子量标记;1~3:pulB扩增产物;CK:不加模板的空白对照
M:DL2000 DNA Marker;1~3:pulB PCR product;CK:PCR without template
图1pulB DNA片段的PCR产物电泳分析
Fig.1Electrophoresis analysis of pulB PCR product
在蛋白质序列上,经用BlastX软件检索GenBank氨基酸数据库发现该基因编码的产物与Thermicanus aegyptius的环麦芽糖糊精酶的氨基酸序列相似性最高,但也仅有54%的相似性(Identities为54%,Positives为69%)。用SMART工具分析该基因编码产物的组件结构,自N端的第133~490位氨基酸为家族GH13糖苷键水解酶α-淀粉酶功能域(图2)。
2.3重组质粒的验证
如图3所示,转化子经双酶切后可切出一大一小两个片段,小片段与预期的大小一致。PCR也可扩增出同样大小的目的片段,证明重组质粒构建成功,命名为pET-pulB。
2.4重组酶PulB的表达和纯化
如图4所示,粗酶液的SDS-PAGE电泳分析显示,在66 kDa位置处有较明显蛋白质特征条带出现,该蛋白质大小与基因序列推测所得的理论分子大小相符,初步认定该蛋白质即为异源表达的普鲁兰酶PulB。酶活力测试结果显示该重组菌具有普鲁兰酶活力,总酶活力最高可达78 U/mL。由此可推断异源表达成功。
上清经Ni-NTA柱进行蛋白质纯化,得到纯度在90%以上的蛋白质(图4)。经过酶活力测定,纯化后的酶具有普鲁兰酶活力,比活力为258 U/mg。
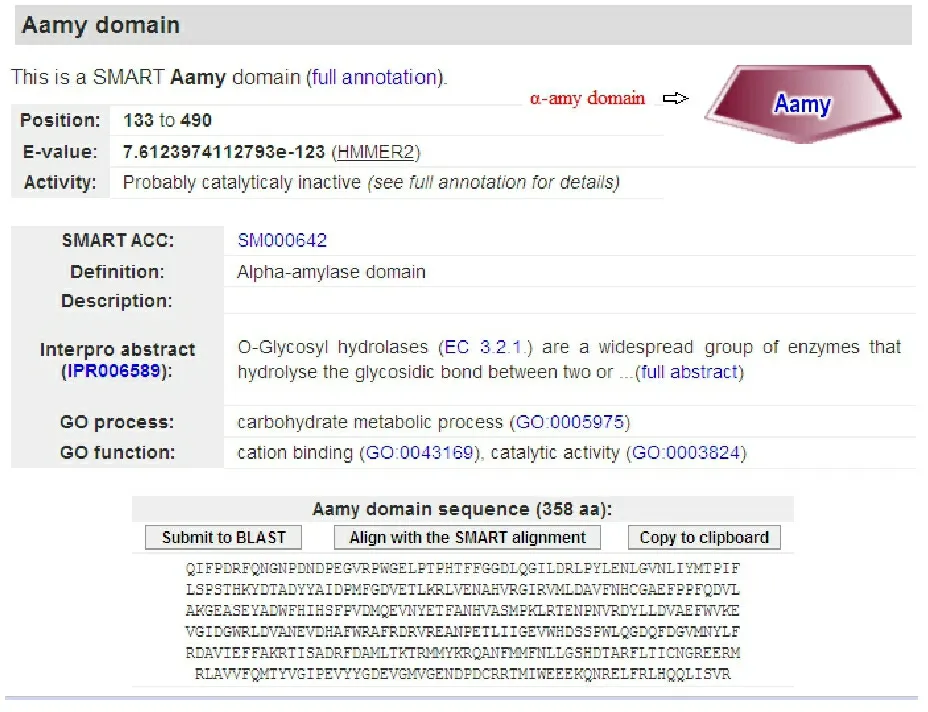
图2pulB的组件结构(红色代表α-amy的功能域)
Fig.2Modular architecture of pulB(The red mark represents α-amy domain)

M1:DL2000 分子量标记;M2:λ/HindⅢ分子量标记;CK:以空质粒为模板的空白对照;1~3:重组质粒pET-pulB;4~9:双酶切验证;10~15:PCR验证
M1:DL2000 DNA Marker;M2:λ/HindⅢ DNA Marker;CK:PCR with pET-22b (+) as template;1~3:recombinant plasmid pET-pulB;4~9:pET-pulB digested withBamHⅠ和HindⅢ; 10~15:pulB PCR product
图3pET-pulB重组质粒验证
Fig.3Verification of recombinant plasmid pET-pulB
2.5重组酶PulB的酶学性质
2.5.1最适反应温度和温度稳定性
由图5a可知,55℃时PulB酶活力达到最高,以该值作100%的酶活力,则45℃时PulB相对酶活力为78%,65℃相对酶活力超过15%。PulB在55℃以下均十分稳定,保温4 h后基本保留80%以上的酶活力,但超过60℃酶失活速度加快,70℃保温4 h后酶活力基本丧失(图5b)。
2.5.2最适反应pH值和pH稳定性
由图6a可知,重组酶PulB最适作用pH值为5.0,当pH值分别为4.6,4.8 和5.4时,PulB的相对酶活力分别为最适pH值条件下的5%、25%和75%,即在较低pH值条件下,PulB酶活力下降比较大。由图6b可知,PulB在pH值为4.5~5.5时均有很好的稳定性,相对酶活力均保持在60%以上。
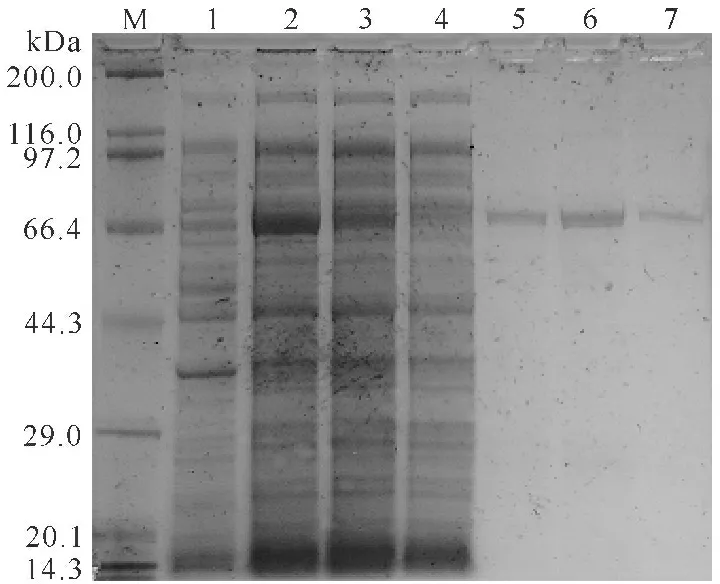
M:蛋白质分子量标记;1:IPTG诱导前的BL21/pET-pulB;2~3:IPTG诱导后的BL21/pET-pulB;4:BL21/pET-22b(+);5~7:纯化的PulB蛋白质
M:Protein Marker;1:Before IPTG induction of BL21/pET-pulB;2~3:After IPTG induction of BL21/pET-pulB;4:BL21/pET-22b(+);5~7:Purificated PulB
图4PulB蛋白表达产物和纯化蛋白质的SDS-PAGE电泳分析
Fig.4SDS-PAGE analysis of PulB expression in BL21 and purificated PulB
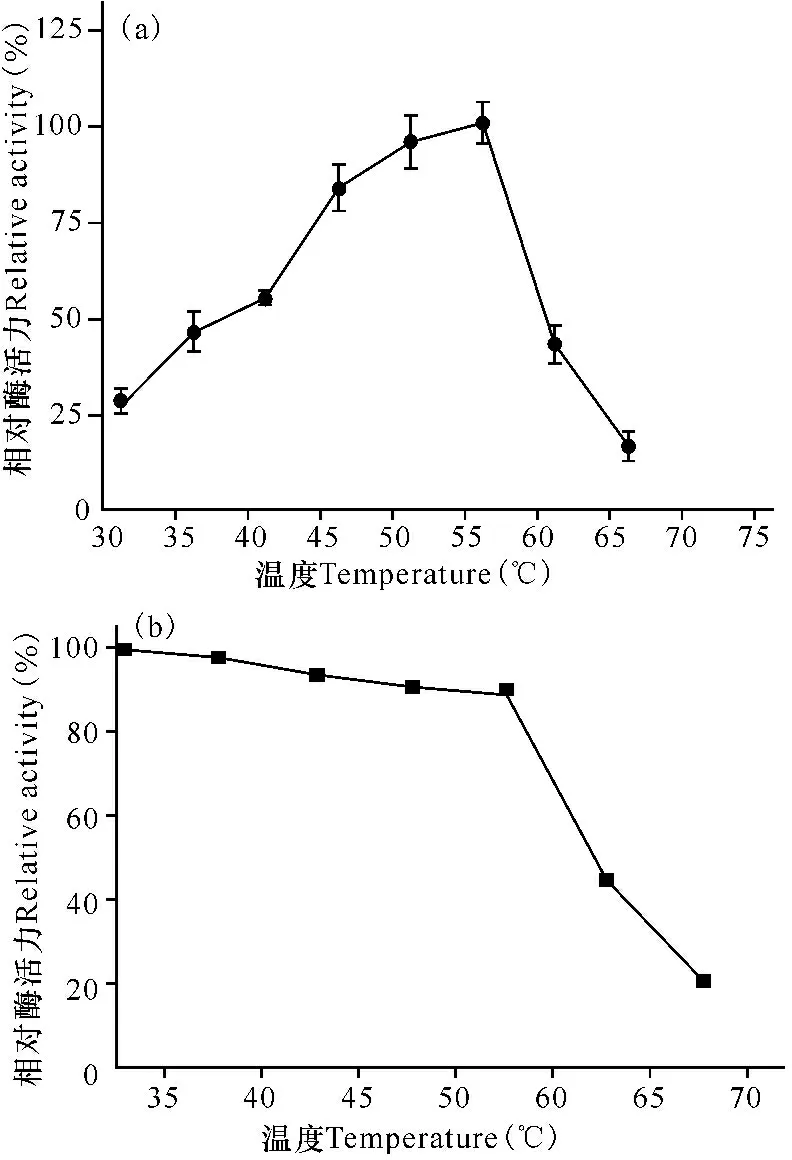
图5PulB的最适反应温度和热稳定性
Fig.5The optimal temperature and thermostability of PulB

图6PulB的最适反应pH值和pH稳定性
Fig.6The optimal pH and pH stability of PulB
2.5.3Km和Vmax的测定
不同来源的酶由于作用的底物不同,其Km值和Vmax也不一样,Km大致范围在10-6到10-1mol/L之间。陆雁[21]从地衣芽孢杆菌中克隆到低温普鲁兰酶,经表达获得的重组酶的Km值为14.29 mmol/L,Vmax值为18.08 μmol·min-1·mg-1。秦艳[22]将长野芽孢杆菌普鲁兰酶基因在大肠杆菌中表达,获得的重组酶的Km值为3.84 mg/mL,Vmax值为31.25 μmol·min-1·min-1。本文研究的来自膨胀芽孢杆菌的重组酶对普鲁兰糖的Km=(16.28±0.03)mg/mL,Vmax=(22.05±0.02)μmol·min-1·mg-1(图7),属于中等水平,这可能与此野生菌的生存环境条件温和,无极端环境条件等因素有关。
2.6金属离子对重组酶PulB水解活性的影响
如图8所示,所有金属离子对PulB都没有明显的促进作用,Zn2+、Pb2+、Co2+、Cu2+对酶活力有明显的抑制作用,其中Cu2+的抑制作用可使酶基本失活,Ba2+对酶活力的抑制作用则相对比较小,而Ca2+、Mg2+、K+、Fe2+、Mn2+则对酶活力几乎不产生影响。
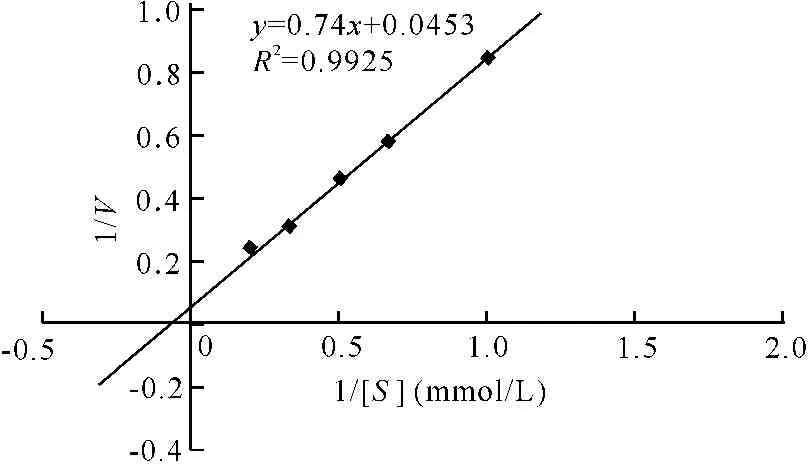

图7 PulB的米氏常数分析
图8不同金属离子对PulB的影响
Fig.8The effect of different metal ions on PulB
2.7重组酶PulB的底物特异性分析
如图9所示,PulB水解普鲁兰糖产生的三糖产物的色谱峰与麦芽三糖标准品的峰的保留时间一致,约为8.003 min,表明PulB通过内切方式水解普鲁兰糖,得到的终产物为三糖。由表1可知,PulB能够水解支链淀粉、可溶性淀粉和α-环糊精,它可水解这些底物中分支链中的α-1,6-糖苷键,也可以将最小单位的支链分解,但不能水解直链淀粉,说明PulB能特异性水解α-1,6-糖苷键,不能作用α-1,4-糖苷键。因此,PulB的催化分类属于Ⅰ型普鲁兰酶。
表1重组普鲁兰酶PulB的底物特异性
Table 1Substrate specificity of recombinant pullulanase PulB

底物*Substrate相对酶活力Relativeactivity(%)比活力Specificactivity(U/mg)普鲁兰糖Pullulan100258.0±0.5可溶性淀粉Solublestarch48125.0±0.4支链淀粉Amylopetin57149.5±0.5α环糊精αcyclodextrins68176.0±0.5直链淀粉Amylose--
注:*,以普鲁兰糖为底物测得的酶活力为100%
Note:*,the activity for pullulan was defined as 100%

图9PulB水解普鲁兰糖产物的HPLC分析
Fig.9HPLC analysis on the products of pullulan hydrolyzed with PulB
3讨论
普鲁兰酶是重要的淀粉水解酶家族成员之一,近年国外对普鲁兰酶的研究比较深入,已从普鲁兰酶结构和功能的关系、普鲁兰酶的定位和分泌机制、结构指导的普鲁兰酶定点突变等方面进行研究[23-24]。国内研究者对酶分子水平上进行改造以获得酶学性质更优的突变酶的研究则比较少,主要是对产酶菌种的筛选及其基因的克隆和异源表达。各国科研人员从来源于不同地区和环境的微生物中获得普鲁兰酶[25-26],但许多酶的酶学性质依然不能满足实际应用的要求,因此需要寻找更多酶学性质更好的产生菌种。
虽然人们已获得产普鲁兰酶的基因工程菌,但普鲁兰酶表达水平及其酶学性质仍有不足,且有的重组质粒在宿主菌中的稳定性不够理想。因此,今后的研究工作一方面可借助启动子改造、信号肽选择、翻译起始区优化等手段构建优良重组质粒,实现重组菌普鲁兰酶的高效表达;另一方面,可以采用定向进化等手段对普鲁兰酶进行分子改造,以进一步改善其酶学特性,如提高酶对底物的亲和力、酶的热稳定性和酸碱耐受性等[27-28]。若能通过上述手段获得耐高温耐酸的普鲁兰酶高产重组菌株,将会对淀粉加工业及相关领域产生深远的影响。
在工业应用上,酶的耐热和耐酸性是重要指标。本研究普鲁兰酶PulB的最适反应温度为55℃,最适反应pH值为5.0,在较窄的酸性pH值范围内(4.5~5.5)较稳定,与目前工业上应用的诺维信公司的嗜酸芽孢杆菌普鲁兰酶的性质比较接近,这些特点也接近目前淀粉加工的生产条件,但仍需进一步改造使其耐酸和耐热性提高才可满足应用的需要。
4结论
本研究采用pET系统实现了T.flagellatus GST4新型普鲁兰酶基因pulB在大肠杆菌中的可溶性表达,发酵液上清酶活力达到78 U/mL,粗酶液经纯化后比活力为258 U/mg,比原始菌的酶活力高15倍。其最适反应温度和pH值分别为55℃和5.0,在低于55℃的温度范围和较窄的酸性范围内(4.5~5.5)具有良好的稳定性,可专一性地水解淀粉和普鲁兰糖中的α-1,6-糖苷键,对普鲁兰糖有较强的底物特异性。酶学性质分析表明,重组普鲁兰酶PulB可用于木薯淀粉生产酒精的水解糖化(作用温度60℃、pH值为4.5~5.5)过程,具有一定的工业化应用价值。
重组普鲁兰酶PulB的DNA序列和GenBank数据库里的任何序列都没有同源性,是尚未报道的新型普鲁兰酶,具有较高的创新性和学术研究价值。通过蛋白质的结构域预测和SMART分析发现,PulB具有淀粉酶的结构域,它可水解普鲁兰糖、支链淀粉、可溶性淀粉和α-环糊精等,属Ⅰ型普鲁兰酶。
参考文献:
[1]HII S L,TAN J S,LING T C,et al.Pullulanase:Role in starch hydrolysis and potential industrial applications[J].Enzyme Res,2012,2012:921362.
[2]莫莉,韦廷宗,闭海,等. 支链淀粉水解酶水解支链淀粉的特异氨基酸分析[J].广西科学,2015,22(1):31-36,43.
MO L,WEI T Z,BI H,et al. Analysis of the key amino acid of amylopectin hydrolase on amylopectin hydrolysis[J].Guangxi Sciences,2015,22(1):31-36,43.
[3]乔宇,丁宏标,王海燕,等.普鲁兰酶的研究进展[J].生物技术进展,2011,1(3):189-194.
QIAO Y,DING H B,WANG H Y,et al.Research advances in pullulanase[J].Current Biotechnology,2011,1(3):189-194.
[4]董桂秀,杨平平,毛朝相,等.普鲁兰酶的研究进展[J].安徽农业科学,2012,40(18):9874-9877.
DONG G X,YANG P P,MAO C X,et al.Research progress on pullulanase[J].Journal of Anhui Agri Sci,2012,40(18):9874-9877.
[5]WALLENEFLS K,BENDER H,RACHED J R.Pullulanase from Aerobacter aeorgenes:Production in a cell-bound state.Purification and properties of the enzyme[J].Biochem Biophys Res Commun,1966,22(3):254-261.
[6]YOSHIYUKI TAKASKAI.Purifications and enzymatic properties of α-amylase and pullulanase from Bacillus cereus var myeoides[J].Agr Biol Chem,1976,40(8):1515-1530.
[7]JENSEN B F,NORMAN B E. Bacillus acidopulluly-ticus pullullanase:Application and regulatory aspects for use in the food industry[J].Process Biochem,1984,19:351-369.
[8]DEWEER P,AMORY A.Pullulanase Producing Microgranisms:5817498[P].1998-10-06.
[9]TOMIMURA E,ZEMAN N W,FRANKIEWICZ J R,et al.Description of Bacillus naganoensis sp.nov.[J].Int J Syst Bacteriol,1990,40(2):123-125.
[10]TOMIMURA E.Thermoduric and Aciduric Pullulan-ase Enzyme and Method for its Production:5055403 [P].1991-10-08.
[11]HYUN H H,ZEIKUS J G.General biochemical characterization of thermostable pullulanase and glucoamylase from Clostridium thermohydrosulfuricum[J].Appl Environ Microbiol,1985,49(5):1168-1173.
[12]韩鹏,周鹏,闫巧娟,等.嗜热枯草芽孢杆菌普鲁兰酶基因的表达与重组酶的性质[J].微生物学通报,2011,38(12):1755-1761.
HAN P, ZHOU P,YAN Q J,et al.Expression of a pullulanase gene from the thermophilic Bacillus subtilis and characterization of therecombinant enzyme[J].Microbiology China,2011,38(12):1755-1761.
[13]杨云娟.普鲁兰酶基因的克隆及其在毕赤酵母中的高效表达[D].昆明:云南师范大学,2005.
YANG Y J.Cloning and Overexpression of Gene Encoding the Pullulanase from Bacillus naganoensis in Pichia pastoris[D].Kunming:Yunnan Normal University,2005.
[14]WANG Q Y,XIE N Z,QIN Y,et al.Tumebacillus flagellatus sp.nov.,an a-amylase/pullulanase-producing bacterium isolated from cassava waste water[J].International Journal of Systematic and Evolutionary Microbiology,2013,63:3138-3142.
[15]SAMBROOK J,RUSSELL D W.分子克隆实验指南[M].3版.北京:科学出版社,2002.
SAMBROOK J,RUSSELL D W.Molecular Cloning:A Laboratory Manual[M].3rd ed.Beijing:Science Press,2002.
[16]LETUNIC I,DOERKS T,BORK P.SMART 6:Recent updates and new developments[J].Nucleic Acids Res,2009,37:229-232.
[17]SCHULTZ J,MILPETZ F,BORK P,et al.SMART,a simple modular architecture research tool:Identification of signaling domains[J].Proc Natl Acad Sci,USA,1998,95(11):5857-5864.
[18]LILIUS G,PERSON M,BU L,et al.Metal affinity precipitation of proteins carrying genetically attached poly histidine tails[J].Eur J Biochem,1991,198(2):499-504.
[19]汪家政.蛋白质技术手册[M].北京:科学出版社,2002.
WANG J Z.The Technical Manuals of Protein[M].Beijing:Science Press,2002.
[20]MALLE D,ITOH T,HASHIMOTO W,et al.Overexpression,purification and preliminary X-ray analysis of pullulanase from Bacillus subtilis strain 168[J].Acta Crystallogr,2006,62:381-384.
[21]陆雁.Bacillus licheniformis普鲁兰酶基因的克隆表达及分子改造[D].南宁:广西大学,2012.
LU Y.Cloning,Expression and Molecular Modification of Pullulanase Gene from Bacillus licheniformis[D].Nanning:Guangxi University,2012.
[22]秦艳.普鲁兰酶突变体及优良酿酒酵母工程菌的构建[D].南宁:广西大学,2014.
QIN Y.Construction of Pullulanase and Genetic Engineering of Saccharomyces cerevisiae Strain[D].Nanning:Guangxi University,2014.
[23]EAST A,MECHALY A E,HUYSMANS G H,et al.Structural basis of pullulanase membrane binding and secretion revealed by X-ray crystallography,molecular dynamics and biochemical analysis[J].Structure,2016,24(1):92-104.
[24]MØLLER M S,ABOU HACHEM M,SVENSSON B,et al.Structure of the starch-debranching enzyme barley limit dextrinase reveals homology of the N-terminal domain to CBM21[J].Acta Crystallogr Sect F Struct Biol Cryst Commun,2012,68(9):1008-1012.
[25]ZHANG Y,LIU Y H,LI Y,et al.Extracellular expression of pullulanase from Bacillus naganoensis in Escherichia coli[J].Ann Microbiol,2013,63(1):289-294.
[26]乔宇.Ⅰ型普鲁兰酶产生菌的筛选及其基因克隆、分子改造和高密度发酵表达研究[D].北京:中国农业科学院,2015.
QIAO Y.Study on Screening of TypeⅠPullulanase Producing Bacteria and its Gene Cloning,Molecular Modification,and Expression in the High Density Fermentation[D].Beijing:Chinese Academy of Agricultural Sciences,2015.
[27]SONG W,NIE Y,MU XQ,et al.Enhancement of extracellular expression of Bacillus naganoensis pullulanase from recombinant Bacillus subtilis:Effects of promoter and host[J].Protein Expr Purif,2016,4(16):30055-30059.
[28]CHEN A,LI Y,NIE J,et al.Protein engineering of Bacillus acidopullulyticus pullulanase for enhanced thermostability using in silico data driven rational design methods[J].Enzyme Microb Technol,2015,78:74-83.
(责任编辑:陆雁)
附件1:pulB基因的核苷酸序列
Attachment 1:The nucleotide sequence of thepulB gene

收稿日期:2016-03-23
作者简介:王青艳(1972-),女,博士,主要从事微生物酶与菌种选育研究。 **通讯作者:黄日波(1958-),男,教授,博士生导师,主要从事酶工程研究,E-mail:rbhuang@gxas.cn。
中图分类号:Q93
文献标识码:A
文章编号:1002-7378(2016)02-0136-10
Cloning,Expression and Enzymatic Characterization of a New TypeⅠ Pullulanase Gene
WANG Qingyan,SHEN Naikun,ZHU Jing,QIN Yan,ZHU Qixia,XIE Nengzhong,LI Yi,HUANG Ribo
(State Key Laboratory of Non-food Biomass and Enzyme Technology,National Engineering Research Center for Non-food Biorefinery,Guangxi Key Laboratory of Biorefinery,Guangxi Academy of Sciences,Nanning,Guangxi,530007,China)
Abstract:【Objective】Screening,cloning,heterologous expression of the pullulanase encoding gene from Tumebacillus flagellatus GST4 in order to obtain efficient expression of a new pullulanase and enhance its activity and thermosatbility.【Methods】Cloning,construction of recombinant and heterologous expression of pullulanase gene in Escherichia coli,purification by Ni-chelating affinity chromatography from cell free culture supernatant and characterization of pullulanase were carried out.【Results】The pullulanase gene pulB was cloned and expressed successfully in E.coli, and the activity of cultural supernatant can reach 78 U/mL.And PulB was purified to homogeneity and the specific activity was 258 U/mg.The optimal temperature of purified PulB is 50℃,its optimal pH value is 5.0 and activity remains stable within the acidic range of pH 4.5~5.5. PulB displayed typical Michaelis-Menten kinetics,where its Kmis(16.28±0.03)mg/mL and Vmaxis(22.05±0.02) μmol·min-1·mg-1,respectively,when used pullulan as substrate.GenBank blast results show that there is no homologous DNA sequences with pulB,and the encoding protein of PulB had the highest identity (54%) with cyclomaltodextrinase from Thermicanus aegyptius.We find that it has amylase structure domain by online SMART searching.The substrate specificity analysis shows that it typically hydrolyze pullulan and amylopectin to produce liner oligosaccharides or maltotriose.【Conclusion】PulB is a new starch/pullulan hydrolase,which has not yet been reported.It can hydrolyze pullulan or amylopectin and belong to type I pullulanase.
Key words:typeⅠpullulanase,Tumebacillus flagellatus,cloning and expression,enzymatic characterization
网络优先数字出版时间:2016-05-17
网络优先数字出版地址:http://www.cnki.net/kcms/detail/45.1075.N.20160517.1545.006.html
*国家自然科学基金项目(31160023,31400079),广西科学研究与技术开发计划项目(桂科合14123001-19,桂科合15104001-1),广西自然科学基金项目(2015GXNSFBA139044),广西“八桂学者”、南宁市特聘专家和留学人员科技活动项目择优资助项目和广西科学院基本科研业务费项目(15YJ22SW01)资助。
