以蛋白激酶C-θ为靶点的免疫抑制剂研究进展
2016-07-18刘俊麟徐翔崔志峰
刘俊麟,徐翔,崔志峰
(浙江工业大学 生物工程学院,浙江 杭州 310032)
以蛋白激酶C-θ为靶点的免疫抑制剂研究进展
刘俊麟,徐翔,崔志峰Δ
(浙江工业大学 生物工程学院,浙江 杭州 310032)
蛋白激酶C(protein kinase C,PKC)至少含有12个亚型,其中PKC的θ亚型(PKC-θ)可参与T细胞受体(T-cell receptor,TCR)和共刺激分子CD28介导的信号整合,调控白细胞介素(interleukin-2,IL-2)的表达和早期T细胞的活化,是自身免疫疾病的主要作用靶点。本文对近年来报道的以PKC-θ为靶点的免疫抑制药物星型孢菌素、Sotrastaurin、噻吩并[2,3-b]吡啶-5-甲腈、吡啶甲腈、氨基嘧啶、AS2521780、甲氧基肉桂醛和单紫杉烯作一综述,旨在为高效、低毒的新型免疫抑制药物的研究提供参考。
蛋白激酶C;PKC-θ;免疫突触;免疫抑制剂
蛋白激酶C(protein kinase C,PKC)是磷脂依赖性的Ser/Thr蛋白激酶。基于PKC对活化剂和辅因子的依赖性,将其分为3个亚族:传统的PKC(conventional PKC,cPKC)亚族(α, βI,βII, γ)需要Ca2+,甘油二酯(diacylglycerol,DAG),磷脂酰丝氨酸(phosphatidylserine,PS)的活化;新型的PKC(novel PKC,nPKC)亚族(ε, δ, θ, η)对Ca2+不敏感,只依赖于DAG和PS;而非典型的PKC(atypical PKC,aPKC)亚族(ζ, ι/λ)对Ca2+和DAG均无依赖性[1]。
PKC的结构(图1)含有4个保守区(C1-C4)和5个可变区
(V1-V5)。PKC的N端调节区差异很大,传统的PKC亚族N端含有假底物区域(PS),DAG结合域(C1a和C1b),Ca2+结合域(C2)。新型的PKC亚族N端含有C2样区域,PS,C1a和C1b区域。非典型的PKC亚族N端没有C2区域,C1区域与其他亚族也不同,不能结合DAG,特殊的PB1(Phox/Bem 1)区域使它可以与其他蛋白质相互作用。PKC的C端保守区含有ATP结合区(C3),底物结合区(C4)[2]。新型PKC亚族中C2样区域不能与Ca2+相结合,但是它可以与活化PKC受体(receptor for activated C kinase,RACK)相互作用,调控PKC的转位现象[3]。
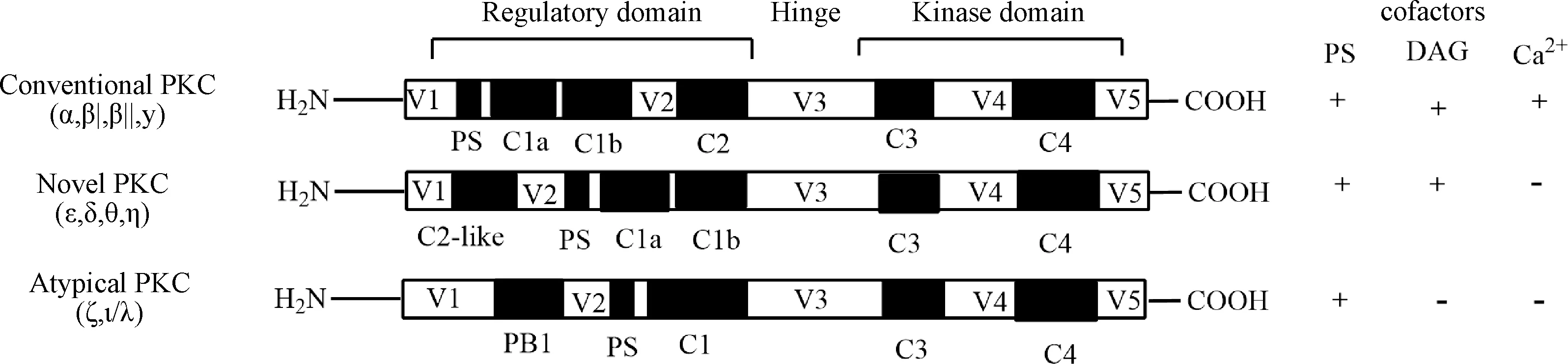
图1 蛋白激酶C的结构[2]Fig.1 Structure of protein kinase C
PKC的θ亚型(PKC-θ)可参与T细胞受体(T-cell receptor,TCR)和共刺激分子CD28介导的信号整合,调控白细胞介素(interleukin-2,IL-2)的表达,对成熟T细胞的活化,增殖和存活发挥重要的作用,是自身免疫疾病的主要作用靶点[4-5]。本文对PKC-θ在T细胞内的调控功能作了简要的介绍,对近年来报道的以PKC-θ为靶点的免疫抑制药物如星型孢菌素、Sotrastaurin、噻吩并[2,3-b]吡啶-5-甲腈、吡啶甲腈、氨基嘧啶、AS2521780、甲氧基肉桂醛和单紫杉烯的结构与作用机理,及其在自身免疫性疾病治疗方面的应用前景研究作一概述。
1 PKC-θ是自身免疫疾病的作用靶点
PKC-θ是PKC家族中新型PKC亚族的成员之一,不依赖Ca2 +,只依赖DAG和PS的激活。活化的PKC-θ可磷酸化特异底物引发胞内信号级联反应,调控多种生命过程和疾病的发生[6]。抗原刺激T细胞后,激活的磷脂酶γ1水解包膜磷脂肌醇并产生DAG,DAG可以与PKC-θ的C1区结合并促进PKC-θ由胞质向胞膜转移,引发PKC-θ的肽链构象改变,N 端假底物区与激酶区解离导致PKC-θ激活[7]。在淋巴细胞特异性蛋白酪氨酸激酶(lymphocyte-specific protein tyrosine kinase,Lck)的参与下,PKC-θ的V3区可以与共刺激分子CD28相结合。与PKC其他亚型相比,PKC-θ最显著的特点是在受到抗原刺激时,它可以易位进入免疫突触(immunological synapse,IS)位点,IS 是指TCR-抗原肽与抗原呈递细胞(antigen-presenting cell,APC)的主要组织相容性复合物( major histocompatibility complex,MHC)识别之后,在T细胞与APC之间形成的接触区。IS由中心超分子激活簇(central supramolecular activation cluster ,cSMAC)和外围超分子激活簇(peripheral supramolecular activation cluster ,pSMAC)组成。TCR/CD28与PKC-θ共同定位于IS,通过信号整合诱导下游信号因子如核转录因子(nuclear factor-κB,NF-κB)、激活蛋白(activator protein 1,AP-1)和活化T细胞核因子(nuclear factor of actiated T cells,NF-AT)调控白细胞介素IL-2的产生[8-10]。
NF-κB是PKC-θ在T细胞内的主要靶标,PKC-θ可磷酸化膜相关鸟苷酸激酶(membrane-associated guanylate kinase,MAGUK)Carma1,Carma1的N端含有caspase募集域(Caspase recruitment domain,CARD)和卷曲螺旋域,磷酸化的Carma1可与Bcl10和Malt1形成低聚物Carma1-Bcl10-Malt1,Malt1可调控转录因子NF-κB的激活,NF-κB核定位序列暴露并从细胞溶质转移进入细胞核内,调控IL-2的基因转录和T细胞活化[11]。激活蛋白AP-1是c-Jun和c-Fos蛋白组成的异源二聚体,PKC-θ可磷酸化STE20相关脯氨酸/丙氨酸丰富激酶(Ste20-related proline/alanine-rich kinase,SPAK),SPAK对AP-1转录因子的激活发挥重要作用[12]。PKC-θ调控NF-AT的转录目前仍有争议,但是,PKC-θ缺陷T淋巴细胞的Ca2+-NFAT通路明显受到部分抑制[13]。
PKC-θ的超表达是自身免疫疾病的标志,如GLK(Germinal center kinase-like kinase,MAPKKKK-3)是PKC-θ的活化剂,当GLK超表达时,可激活PKC-θ,刺激IKK(inhibitor of nuclear factor kappa-B kinase,IKK),导致系统性红斑狼疮的发生,并且,在类风湿性关节炎患者的外周血T细胞中GLK的表达量明显高于健康受试者[14]。此外,PKC-θ的活性受到抑制可改善患者的疾病状态,如在Th1(T Helper Type 1,Th1)依赖性抗原诱导的关节炎模型中,PKC-θ缺陷性小鼠与野生型小鼠相比,其骨破坏和关节软骨的损害程度明显较轻,可能与CD4+T细胞内细胞因子IFN-γ,IL-2,IL-4的表达密切相关[15]。在异基因骨髓移植模型中,PKC-θ过表达可促进移植物抗宿主疾病(graft-versus-host-disease,GVHD),PKC-θ受到抑制后能够明显改善患者器官移植后的排斥反应[16]。因此,PKC-θ参与TCR/CD28 诱导的T细胞活化信号级联反应,对于成熟T淋巴细胞的增殖和活化具有重要意义,PKC-θ是一个新型的自身免疫疾病的治疗靶点。
2 以PKC为靶标的免疫抑制剂
2.1 星型孢菌素类化合物 星型孢菌素(Staurosporine) 是从链霉菌(Streptomycesstaurosporeus)的代谢物中提取得到的吲哚[2,3-α]咔巴唑类生物碱(图2-A),可有效的抑制PKC激酶的活性,IC50为2.7 nmol/L[17]。以星型孢菌素作为先导物,合成了大量的吲哚咔巴唑和吲哚马来酰亚胺类物质,其中典型化合物为Sotrastaurin(STN,AEB071)(图2-B)[18]。

图2 星型孢菌素类化合物Fig.2 Staurosporine and its derivatives
STN是由诺华制药(Novartis)公司研发,化学名为3-(1H-吲哚-3-基)-4-[2-(4-甲基哌嗪-1-基)喹唑啉-4-基]-吡咯-2,5-二酮,STN的喹唑啉基和吲哚部分与PKC的ATP结合位点紧密结合,可抑制PKC-α,PKC-βⅠ,PKC-δ,PKC-ε,PKC-η,PKC-θ亚型,IC50分别为2.1 nmol/L,2.0 nmol/L,1.3 nmol/L,6.2 nmol/L,6.1 nmol/L,1.0 nmol/L。STN抑制小鼠和人体混合T淋巴细胞反应(Mixed lymphocyte reactions,MLR)的IC50分别为128 nmol/L和34 nmol/L,抑制效率相当于环孢素的1/3[18]。STN抑制TCR/CD28介导的Jurkat T细胞活化后IL-2的表达,IC50为54 nmol/L[19]。
STN是以PKC为靶点的免疫抑制剂,目前已用于自身免疫性疾病临床研究。STN治疗银屑病疗效显著,随机口服25,100,200和300 mg的STN,每天2次,服用2周后,临床和组织学检测显示,服用剂量为300 mg的患者第14天,银屑病面积严重程度指数(psoriasis area severity index,PASI)下降了69%,且不良反应较少,服用安慰剂组的患者PASI仅下降了5.3%[20]。但是,STN用于肾移植和肝脏移植存在严重的不良反应。双盲、随机和多中心的肾移植临床Ⅱ期研究显示STN并未具有优于FK506的治疗效果[21]。其临床Ⅱ期肝脏移植研究仅仅实施了6个月就被迫终止[22]。STN能否用于器官移植治疗令人担忧。
2.2 噻吩并[2,3-b]吡啶-5-甲腈类化合物 惠氏制药(Wyeth)公司Nathan等人以噻吩并[2,3-b]吡啶为先导物合成2-链烯基噻吩并[2,3-b]吡啶-5-甲腈(图3-A),其抑制PKC-θ的IC50为3.8 nmol/L。动物细胞试验显示[23],该物质抑制抗CD3/CD28抗体诱导的野生型小鼠T淋巴细胞IL-2产生,IC50为170 nmol/L,抑制PKC-θ缺陷型小鼠T淋巴细胞IL-2产生,IC50>1000 nmol/L。因此,认为它的确阻碍T淋巴细胞内PKC-θ激酶信号通路。但是,该化合物水溶性较差,并且也抑制酪氨酸激酶Lyn的活性[23]。Lyn参与B细胞抗原受体(B cell antigen receptor,BCR)介导的信号转导通路,Lyn活性受到抑制后,降低了外周血B淋巴细胞终末端分化,对治疗自身免疫疾病极为不利[24],因此,该物质并未做相关的药物研究。
Wu等[25]合成新的噻吩并[2,3-b]吡啶类化合物,4-(4-甲基-1H-吲哚-5-氨基)-2-苯基噻吩并[2,3-b]吡啶-5-甲腈(图3-B),其C-4位为4-甲基-5-吲哚基胺,C-2位为5-甲氧基-2-(二甲基氨基)甲基苯基。它抑制PKC-θ, PKC-β,PKC-η,PKC-ε,PKC-δ,IC50分别为16、22、360、95、130 nmol/L,而抑制Src激酶家族(Src family kinase,SFK)Lck,Src,Hck,Fyn等的 IC50均大于5 μmol/L。但是,其在小鼠和人体肝脏内稳定性不好,相关的免疫抑制试验未见报道。
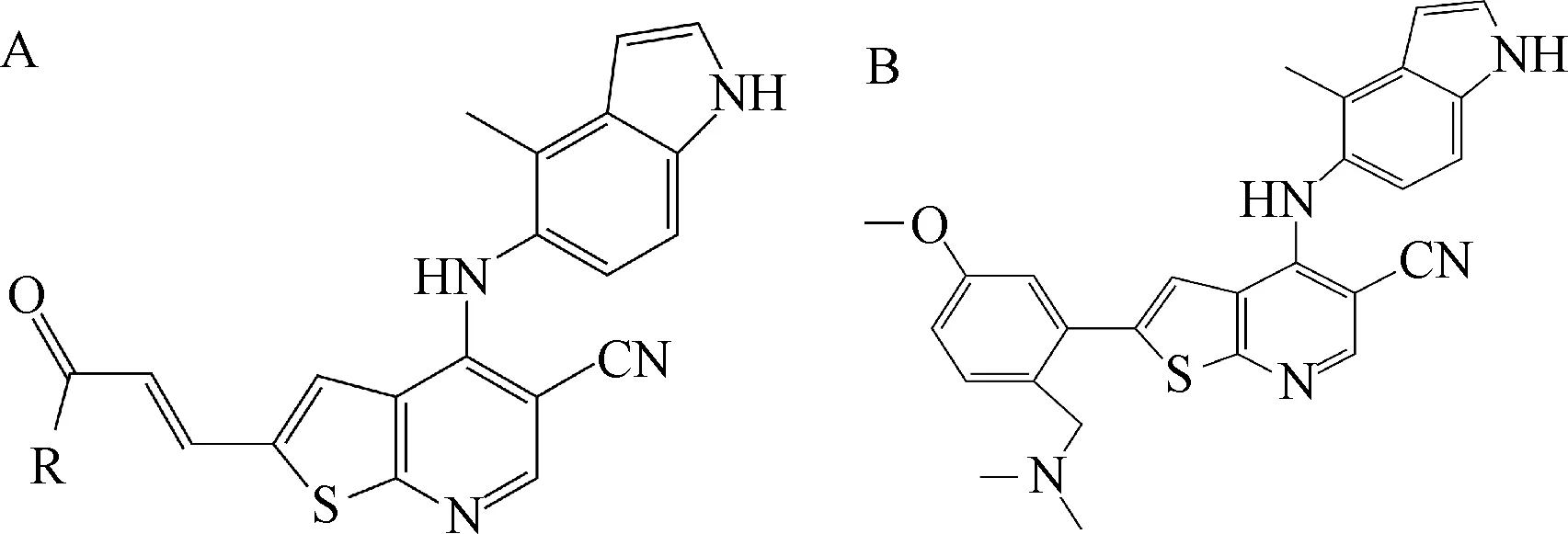
图3 噻吩并[2,3-b]吡啶-5-甲腈类化合物Fig.3 Thieno [2,3-b]pyridine-5-carbonitrile derivatives
2.3 吡啶甲腈类化合物 Joan等人报道人工合成的第一代3-吡啶腈类PKC-θ抑制剂(图4-A),其C-4位为4-甲基吲哚-5-氨基基团, C-5位为5-[(4-甲基哌嗪-1-基)甲基]-2-呋喃[26]。它抑制PKC-θ,PKC-η,PKC-ε,PKC-β的IC50分别为4.5 nmol/L,140 nmol/L,12 nmol/L,2.5 μmol/L。抑制野生型小鼠T淋巴细胞IL-2产生的IC50为86 nmol/L,抑制PKC-θ缺陷型小鼠T淋巴细胞IL-2产生的IC50为1900 nmol/L。因此,这是一个专一性强的PKC-θ抑制剂[27]。
Prashad等人以双环杂芳环作为取代基,合成第二代3-吡啶腈类PKC-θ抑制剂(图4-B),其C-4位为4-甲基吲哚-5-氨基,C-5位为5-[(4-甲基哌嗪-1-基)甲基]-1-苯并呋喃-2-基。它抑制 PKC-θ,PKC-β,PKC-η,PKC-ε,PKC-ζ的IC50分别为0.28 nmol/L, 2.5 μmol/L,33 nmol/L,2.5 nmol/L,>100 μmol/L。抑制野生型小鼠T淋巴细胞IL-2产生的IC50为21 nmol/L,抑制PKC-θ缺陷型小鼠T淋巴细胞IL-2产生的IC50为880 nmol/L。它可逆性的抑制人肝微粒细胞色素P450活性的IC50大于10 μmol/L,口服生物利用率为35%,是一个理想的PKC-θ抑制剂,但是其药代动力学研究未公布[28]。
Boschelli等[29]对5-乙烯基芳基-3-吡啶腈结构优化后得到新一代的3-吡啶腈类PKC-θ抑制剂,其C-5位为(E)-2-{6-[(4-甲基哌嗪-1-基甲基]吡啶-2-基}乙烯基(图4-C),抑制PKC-θ的IC50为1.1 nmol/L。此外,它抑制小鼠和人T淋巴细胞白介素IL-2产生,IC50分别为34 nmol/L和500 nmol/L,其代谢和溶解度稳定,有望成为潜在的以PKC-θ为靶点的免疫抑制药物。
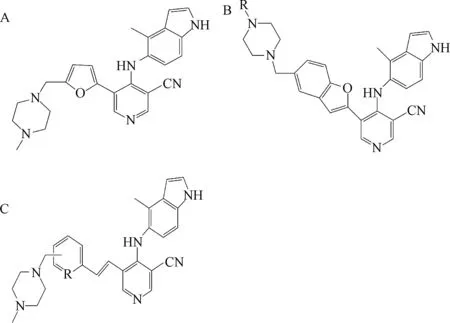
图4 吡啶甲腈类化合物Fig.4 Pyridinecarbonitrile derivatives
2.4 氨基嘧啶类化合物 2,4-二氨基-5-氟嘧啶类化合物(图5-A)是由Rigel报道的PKC-θ抑制剂,但它对细胞色素P4503A4(CYP3A4)也表现出时间依赖性抑制(Time-dependent inhibition,TDI),而且还是P-糖蛋白(P-glycoprotein,P-gp)的底物。最近,Kunikawa等[30]以2,4-二氨基-5-氟嘧啶为先导物,合成一系列2,4-二氨基-5-氟嘧啶类的PKC-θ抑制剂,其中代表化合物(图5-B),化学名为N2-[3-(4-氯-1H-吡唑-1-基)苯基]-5-氟-N4-{[(3S)-1-甲基-3-基]甲基}嘧啶-2,4-二胺,抑制PKC-θ的IC50为3.0 nmol/L,而且特异性提高,对CYP3A4无明显的抑制效应,也非P-糖蛋白(P-gp)的底物。化合物5-B的免疫抑制活性尚未公布。
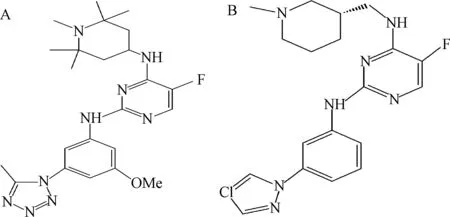
图5 氨基嘧啶类化合物Fig.5 Aminopyrimidines derivatives
2.5 AS2521780 日本Fukahori等[31]首次发现AS2521780(图6)能够抑制人体PKC,抑制PKC-θ的IC50为 0.48 nmol/L,是PKC其他亚型抑制效率的30倍,并且对酪氨酸激酶和丝氨酸/苏氨酸激酶均无明显的抑制活性。体外动物细胞实验显示,AS2521780抑制TCR信号转导通路中IL-2产生,IC50为14.0 nmol/L,抑制CD3/CD28诱导的人体T细胞增殖,IC50为17.0 nmol/L。AS2521780抑制鼠脾细胞和食蟹猴外周血淋巴细胞(peripheral blood lymphocytes,PBLs)细胞因子IL-2的产生,IC50分别为8.9 nmol/L和10.5 nmol/L。小鼠口服AS2521780分别为3, 10 和30 mg/kg,以剂量依赖性的方式抑制伴刀豆球蛋白A(Concanavalin A,ConA)诱导的细胞因子IL-2, IFN-γ,TNF-α和IL-17A的产生。小鼠诱导佐剂性关节炎模型表明,30 mg/kg的AS2521780几乎完全改善了小鼠足肿胀现象,无毒副作用。
小鼠异位心脏移植模型显示,AS2521780单药治疗能够延长小鼠移植存活时间,口服30 mg/kg的AS2521780,心脏移植小鼠存活时间为20 d,同等剂量的STN(蛋白激酶非特异性免疫抑制药物)治疗的小鼠移植存活时间仅为14 d,可见AS2521780与STN相比,疗效更好。1 mg/kg的AS2521780分别与0.02 mg/kg的 FK506,15 mg/kg的霉酚酸酯联合治疗心脏移植,移植存活时间与单药治疗相比,可延长13 d。此外,非人灵长类NHP肾移植模型也出现同样的结果,肾组织病理学研究结果显示FK506治疗肾移植后,6/7的动物患有IIA, IIB和 III 型急性T细胞调节的排斥反应(T cell-mediated rejection,TCMR),而用AS2521780治疗后,5/6的动物的TCMR并未超过I型。与FK506相比,AS2521780治疗肾移植安全性更好,而且动物体重和血浆肝酶(如天冬氨酸转氨酶、丙氨酸转氨酶、碱性磷酸酶)的水平均无明显升高。从以上实验均可看出,AS2521780确实能显著的延长动物器官移植存活时间[32]。但是,其临床治疗人体自身免疫性疾病有待研究。
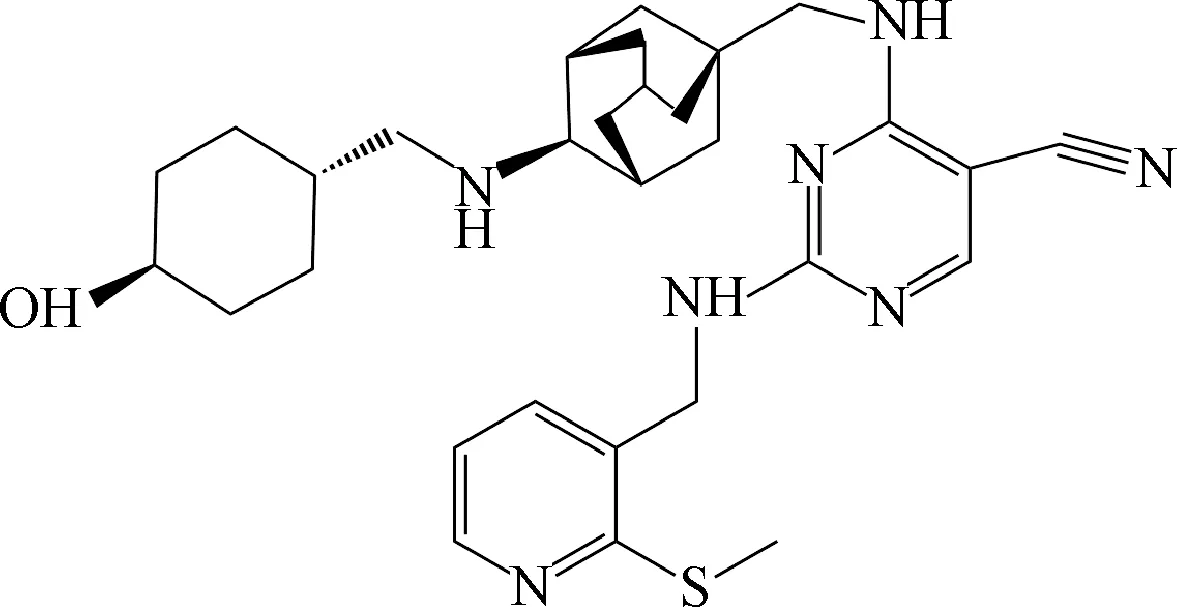
图6 AS2521780Fig.6 AS2521780
2.6 甲氧基肉桂醛类化合物 4-羟基肉桂酸(4-hydroxycinnamic acid,HCA)是T细胞抑制剂,其衍生物4-羟基-3-甲氧基肉桂醛(4-hydroxy-3-methoxycinnamaldehyde,4H3MC,图7)具有抗真菌活性,并能够抑制黑色素瘤细胞黑色素的产生。最近,Akber等[33]研究发现,4H3MC还具有免疫抑制的活性,能够显著性的抑制Jurkat T细胞和人外周血白细胞(peripheral blood leukocytes,PBLs)细胞因子IL-2的产生,其IC50分别为2.5 μmol/L和2.8 μmol/L,其对T细胞的抑制活性是HCA的5倍左右。研究显示,4H3MC明显的抑制了PKC激酶的活性及T细胞中PKC-θ的磷酸化作用,对TCR介导的膜-近端信号通路如Zap70的磷酸化作用效果不明显。因此,认为4H3MC通过PKC-θ途径抑制T细胞的活性。
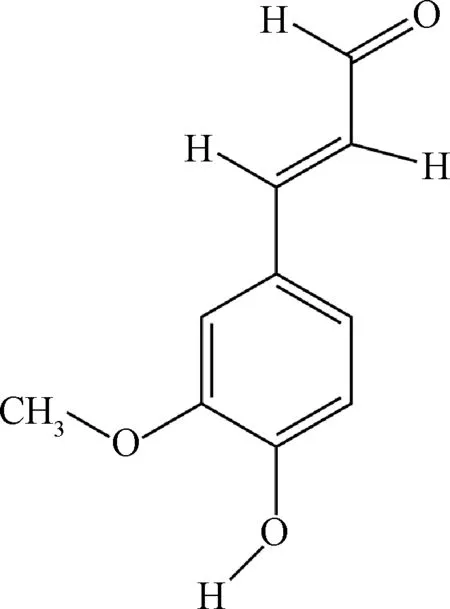
图7 4-羟基-3-甲氧基肉桂醛Fig.7 4-hydroxy-3-methoxycinnamaldehyde
2.7 单紫杉烯 单紫杉烯(Aplotaxene)的结构式为C17H28,化学名为(8Z,11Z,14Z)-十七-1,8,11,14,四烯,是土木香根部提纯获得的香精油的主要成分(图8)。土木香,为菊科旋覆花属,多年生草本植物,可供药用,其根可治疗哮喘,咳嗽,肺结核,消化不良,以及传染病和寄生虫病等其他各种疾病[34]。已报道土木香根部提纯物具有抗菌,抗肿瘤和抗恶性细胞增殖的活性[35]。
Na等[36]首次发现土木香根部提纯物单紫杉烯具有免疫抑制活性,单紫杉烯以浓度依赖性的方式抑制Jurkat T细胞的细胞因子IL-2的表达,IC50为5-10 μmol/L,3 h内抑制活性最强,6 h内活性稳定。CD69是早期T细胞活化后即可检测到的表面分子,研究发现,单紫杉烯使CD69的表达量显著降低,单紫杉烯对人外周血白细胞PBLs的细胞因子IL-2的表达同样有抑制作用。并且,其最佳抑制浓度对细胞凋亡和坏死并无影响,初步表明单紫杉烯并非是诱导T细胞凋亡而抑制T细胞活化。对单紫杉烯免疫抑制机理的研究发现,在抗CD3/CD28和PMA/A23187处理的T细胞中,单紫杉烯显著性的抑制了T细胞内PKC-θ的磷酸化。在T细胞与B细胞结合试验中,共聚焦显微镜观察显示单紫杉烯不仅抑制了PKC-θ的磷酸化,并阻碍了PKC-θ在免疫突触位点cSMAC区的积累。此外,单紫杉烯对PKC-α,PKC-ε, PKC-δ的磷酸化作用无影响。但是,PKC-ζ的磷酸化受到抑制,并且发现,PKC-ζ与PKC-θ是一种物理性的相互作用,在PKC-θ的下游发挥作用。实验显示,单紫杉烯具有以PKC-θ为靶标的免疫抑制作用。
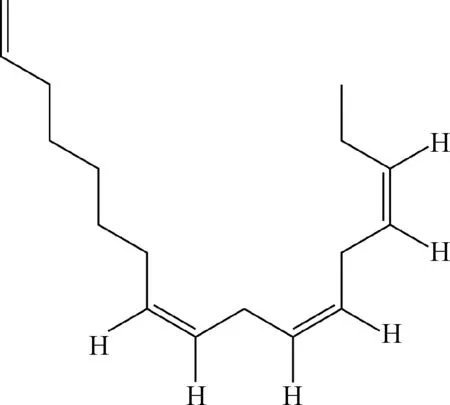
图8 单紫杉烯Fig.8 Aplotaxene
3 结语
PKC-θ是新型的PKC亚型之一,不依赖于Ca2+,PKC-θ可参与调控TCR和共刺激分子CD28介导的信号整合,调控IL-2的表达和早期T细胞的活化,是自身免疫疾病的新型的作用靶点[4-5]。星型孢菌素是第一个报道的以PKC为靶标的免疫抑制剂,其衍生物STN是诺华制药公司开发的治疗银屑病的可选药物[20]。诺华制药公司还报道了大量人工合成的PKC-θ抑制剂,为其用于免疫疾病治疗做了大量的研究。AS2521780是迄今为止报道的以PKC-θ为靶点的免疫抑制剂中抑制活性最高,特异性最好的。在小鼠关节炎模型、小鼠异位心脏移植模型和非人灵长类NHP肾移植模型中,AS2521780都显示出低毒、高效的特性,这些实验数据可以为AS2521780的临床研究提供重要的参考[31-32]。药用植物来源的单紫杉烯具有明确的以PKC-θ为靶标的免疫抑制作用,药用植物提取物副作用小,在器官移植和自身免疫疾病的辅助治疗方面可能会有举足轻重的作用[36]。因此,以PKC-θ为靶点的免疫抑制药物的研究具有较好的发展前景。
[1] Igumenova TI. Dynamics and Membrane Interactions of Protein Kinase C[J].Biochemistry, 2015, 54(32): 4953-4968.
[2]Lim PS, Sutton CR, Rao S. Protein kinase C in the immune system: from signalling to chromatin regulation[J].Immunology, 2015, 146(4): 508-522.
[3]Schechtman D, Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction[J].Oncogene, 2001, 20(44): 6339-6347.
[4]Chand S, Mehta N, Singh Bahia M, et al. Protein kinase C-theta inhibitors: a novel therapy for inflammatory disorders[J].Curr Pharm Des, 2012, 18(30): 4725-4746.
[5]Hage-Sleiman R, Hamze AB, Reslan L, et al. The Novel PKC θ from Benchtop to Clinic[J].J Immunol Res, 2015(2015): 348798.
[6]Marsland BJ, Kopf M. T-cell fate and function: PKC-θ and beyond[J].Trends Immunol, 2008, 29(4): 179-185.
[7]Majhi A, Rahman GM, Panchal S, et al . Binding of curcumin and its long chain derivatives to the activator binding domain of novel protein kinase C[J].Biorg Med Chem, 2010, 18(4): 1591-1598.
[8]Pfeifhofer-Obermair C, Thuille N, Baier G. Involvement of distinct PKC gene products in T cell functions[J].Front Immunol, 2012(3): 220.
[9]Sedwick CE, Altman A. Perspectives on PKC theta in T cell activation[J].Mol Immunol, 2004, 41(6-7): 675-686.
[10]Zhang EY, Kong KF, Altman A. The yin and yang of protein kinase C-theta (PKC theta): a novel drug target for selective immunosuppression[J].Adv Pharmacol, 2013(66): 267-312.
[11]Zanin-Zhorov A, Dustin ML, Blazar BR. PKC-θ function at the immunological synapse: prospects for therapeutic targeting[J].Trends Immunol, 2011, 32(8): 358-363.
[12]Li Y, Hu J, Vita R, et al. SPAK kinase is a substrate and target of PKCθ in T‐cell receptor‐induced AP‐1 activation pathway[J].EMBO J, 2004, 23(5): 1112-1122.
[13]Pfeifhofer C, Kofler K, Gruber T, et al. Protein kinase C θ affects Ca2+mobilization and NFAT activation in primary mouse T cells[J].J Exp Med, 2003, 197(11): 1525-1535.
[14]Chen YM, Chuang HC, Lin WC, et al. Germinal Center Kinase-like Kinase Overexpression in T Cells as a Novel Biomarker in Rheumatoid Arthritis[J].Arthritis Rheum, 2013, 65(10): 2573-2582.
[15]Healy AM, Izmailova E, Fitzgerald M, et al. PKC-θ-deficient mice are protected from Th1-dependent antigen-induced arthritis[J].J Immunol, 2006, 177(3): 1886-1893.
[16]Valenzuela JO, Iclozan C, Hossain MS, et al. PKCθ is required for alloreactivity and GVHD but not for immune responses toward leukemia and infection in mice[J].J Clin Invest, 2009, 119(12): 3774.
[17]Tamaoki T, Nomoto H, Takahashi I, et al. Staurosporine, a potent inhibitor of phospholipid Ca++dependent protein kinase[J].Biochem Biophys Res Commun, 1986, 135(2): 397-402.
[18]Wagner Jr, von Matt P, Sedrani R, et al. Discovery of 3-(1 H-indol-3-yl)-4-[2-(4-methylpiperazin-1-yl) quinazolin-4-yl] pyrrole-2, 5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes[J].J Med Chem, 2009, 52(20): 6193-6196.
[19]Wagner J, von Matt P, Faller B, et al. Structure-activity relationship and pharmacokinetic studies of sotrastaurin (AEB071), a promising novel medicine for prevention of graft rejection and treatment of psoriasis[J].J Med Chem, 2011, 54(17): 6028-6039.
[20]Skvara H, Dawid M, Kleyn E, et al. The PKC inhibitor AEB071 may be a therapeutic option for psoriasis[J].J Clin Invest, 2008, 118(9): 3151.
[21]Friman S, Arns W, Nashan B, et al. Sotrastaurin, a Novel Small Molecule Inhibiting Protein‐Kinase C: Randomized Phase II Study in Renal Transplant Recipients[J].Am J Transplantation, 2011, 11(7): 1444-1455.
[22]Pascher A, De Simone P, Pratschke J, et al. Protein kinase C inhibitor sotrastaurin in de novo liver transplant recipients: A randomized phase II trial [J].Am J Transplantation, 2015, 15(5): 1283-1292.
[23]Tumey LN, Boschelli DH, Lee J, et al. 2-Alkenylthieno [2,3-b] pyridine-5-carbonitriles: Potent and selective inhibitors of PKCtheta[J].Bioorg Med Chem Lett, 2008, 18(15): 4420-4423.
[24]Nishizumi H, Taniuchi I, Yamanashi Y, et al. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice [J].Immunity, 1995, 3(5): 549-560.
[25]Wu B, Boschelli DH, Lee J, et al. Second generation 4-(4-methyl-1H-indol-5-ylamino)-2-phenylthieno[2,3-b]pyridine-5-carbonitrile PKCtheta inhibitors[J].Bioorg Med Chem Lett, 2009, 19(3): 766-769.
[26]Subrath J, Wang D, Wu B, et al. C-5 Substituted heteroaryl 3-pyridinecarbonitriles as PKCtheta inhibitors: Part I[J].Bioorg Med Chem Lett, 2009, 19(18): 5423-5425.
[27]Boschelli DH, Wang D, Prashad AS, et al. Optimization of 5-phenyl-3-pyridinecarbonitriles as PKCtheta inhibitors[J].Bioorg Med Chem Lett, 2009, 19(13): 3623-3626.
[28]Prashad AS, Wang D, Subrath J, et al. C-5 substituted heteroaryl-3-pyridinecarbonitriles as PKC theta inhibitors: part II[J].Bioorg Med Chem Lett, 2009, 19(19): 5799-5802.
[29]Boschelli DH, Subrath J, Niu C, et al. Optimization of 5-vinylaryl-3-pyridinecarbonitriles as PKC theta inhibitors[J].Bioorg Med Chem Lett, 2010, 20(6): 1965-1968.
[30]Kunikawa S, Tanaka A, Mukoyoshi K, et al. Optimization of 2,4-diamino-5-fluoropyrimidine derivatives as protein kinase C theta inhibitors with mitigated time-dependent drug-drug interactions and P-gp liability[J].Bioorg Med Chem, 2015, 23(13): 3269-3277.
[31]Fukahori H, Chida N, Maeda M, et al. Effect of AS2521780, a novel PKCtheta selective inhibitor, on T cell-mediated immunity[J].Eur J Pharmacol, 2014(745): 217-222.
[32]Fukahori H, Chida N, Maeda M, et al. Effect of novel PKC theta selective inhibitor AS2521780 on acute rejection in rat and non-human primate models of transplantation[J].Int Immunopharmacol, 2015, 27(2): 232-237.
[33]Akber U, Na BR, Ko YS, et al. Phytocomponent 4-hydroxy-3-methoxycinnamaldehyde ablates T-cell activation by targeting protein kinase C-theta and its downstream pathways[J].Int Immunopharmacol, 2015, 25(1): 130-140.
[34]Konishi T, Shimada Y, Nagao T, et al. Antiproliferative sesquiterpene lactones from the roots of Inula helenium[J].Biol Pharm Bull, 2002, 25(10): 1370-1372.
[35]Dorn DC, Alexenizer M, Hengstler JG, et al. Tumor cell specific toxicity of Inula helenium extracts[J].Phytother Res, 2006, 20(11): 970-980.
[36]Na BR, Kim HR, Kwon MS, et al. Aplotaxene blocks T cell activation by modulation of protein kinase C-theta-dependent pathway[J].Food Chem Toxicol, 2013(62): 23-31.
(编校:谭玲)
Progresses in the research of immunosuppressor which take protein kinase C-θ as target
LIU Jun-lin, XU Xiang, CUI Zhi-fengΔ
(College of Biological Engineering, Zhejiang University of Technology, Hangzhou 310032, China)
Protein kinase C (PKC) is consisted of at least 12 subunits, and its theta subunit (PKC-θ) was found can participate in the signal integration of T cell receptor (TCR) and co-stimulatory molecules CD28, and regulate the expression of Interleukin-2 (IL-2) and activation of early stage T cells, is the primary target of autoimmune disease. This review summarize the immunosuppressive drugs which take the PKC-θ as their target, including staurosporine, sotrastaurin, thieno[2,3-b]pyridine-5-carbonitriles, pyridine carbonitrile, amino pyrimidine, AS2521780, methoxycinnamaldehyde and aplotaxene, aim at providing reference of research for new immunosuppressive drugs with high efficiency and low toxicity.
protein kinase C; PKC-θ; immunological synapse; immunosuppressor
刘俊麟,女,硕士,研究方向:酶抑制剂的筛选,E-mail:liujunlin1991@163.com;崔志峰,通信作者,男,博士,教授,研究方向:酶抑制剂研究, E-mail:zfcui@zjut.edu.cn。
R979.1
A
10.3969/j.issn.1005-1678.2016.03.60
