Gitelman综合征:附1例报道并文献复习
2016-07-08陈芬琴王涤非刘珂吴坤艳张艳玲赵岩
陈芬琴,王涤非,刘珂,吴坤艳,张艳玲,赵岩
(中国医科大学附属第一医院老年病科,沈阳110001)
Gitelman综合征:附1例报道并文献复习
陈芬琴,王涤非,刘珂,吴坤艳,张艳玲,赵岩
(中国医科大学附属第一医院老年病科,沈阳110001)
摘要Gite1man综合征是一种少见性遗传性疾病,本文通过回顾性分析1例Gite1man综合征患者临床表型特征及基因测序,发现Gite1man综合征可能存在SLC12A3基因外的突变,需要更多的基因筛查研究加以证实。
关键词Gite1man综合征;低血钾;低尿钙;SLC12A3
网络出版地址
网络出版时间:
Gite1man综合征(Gite1man syndrome,GS)是一种常染色体隐性遗传性肾小管疾病,也被称为家族性低钾低镁血症。1966年由Gite1man首次发现,其病因为编码肾远曲小管上皮细胞上噻嗪类敏感性Na/C1协同转运(Na-C1 cotransporter,NCCT)基因突变所致。临床表现为顽固性低钾血症、低镁血症、低尿钙、血压正常或降低,同时可伴低氯性碱中毒,肾素-血管紧张素-醛固酮水平升高,部分患者醛固酮水平也可能为正常,肾脏病理检查可见肾小球旁器增生[1]。GS的发病率较低且临床认识不足,基因分析更是贫乏。本研究总结了我科收治的1例 GS患者的临床资料及基因序列分析结果,并参阅国内外相关文献分析本病基因型与表型之间的关系。
1 临床资料
女性患者,33岁,以“乏力10余年,双手抽搐半个月”为主诉于2015年7月入院。患者于10余年前出现乏力,8年前行剖腹产手术时被告知低血钾,对症补钾后未系统诊治。半个月前无明显诱因出现双手抽搐,伴麻木无力感,急查血钾为2.49 mmo1/L,给予静脉及口服补钾治疗,上述症状好转,但复查血钾波动在2.38~3.20 mmo1/L之间,多次测血压正常,为明确低钾原因收入我科。患者病来无恶心呕吐,无腹痛腹泻病史,无多尿多饮史,否认服用利尿剂及其他药物史,饮食及睡眠可,发育正常,育有1女,家族中无类似疾病史,父母亲均健康,无其他兄弟姐妹,1女健康。
患者身高157 cm,体质量47.5 kg,体质量指数19.27 kg/m2,血压106/69 mmHg,心、肺、腹部无异常,Trousseau征及Chvostek征阴性。入院后多次查血钾为2.61~3.21 mmo1/L(参考值3.5~5.3 mmo1/L),血镁0.36~0.47 mmo1/L(参考值0.78~1.28 mmo1/L),血氯94.3~101.1 mmo1/L(参考值99~110 mmo1/L),血碳酸氢盐26.3~31.5 mmo1/L(参考值22~29 mmo1/L),血钠138.4~141.4 mmo1/L(参考值137~147 mmo1/L),血钙2.19~2.53 mmo1/L(参考值2.17~2.57 mmo1/L)。24 h尿量750~1 430 mL,24 h尿钾分别为88.61、91.44、71.52 mmo1,24 h尿镁分别为3.30、3.16、3.44 mmo1,24 h尿氯分别为156.86、216.97、172.26 mmo1,24 h尿钠分别为125.19、197.86、146.34 mmo1,24 h尿钙分别为1.73、1.23、1.28 mmo1。尿钙/尿肌酐分别为0.21、0.10、0.07。血气分析:pH7.438,PaO279.6 mmHg,PaCO235.8 mmHg,HCO3-29.4 mmo1/L,剩余碱(base excess,BE)6.3 mmo1/L。肾素—血管紧张素—醛固酮检测结果见表1,甲状腺功能、肝肾功能、甲状旁腺激素(parathyroid hormone,PTH)、肾上腺皮质激素系列、风湿抗体系列和血沉均未见明显异常。
肾上腺增强CT:左侧肾上腺内侧支增粗,密度均匀。腹部彩超:脂肪肝。心电图:轻度ST改变。
基因序列检测回报有6处突变,其中外显子1 c.-213C>G,c.142C>T,这2处突变均为非编码区突变;外显子2 c.366A>G,导致A1a122A1a,不过此为同义突变;外显子2,21,23有3处内含子突变(表2,图1)。
患者存在低血钾、低血氯、低血镁,血压正常,肾素活性增高,低尿钙,尿钙/肌酐<0.2,临床诊断“Gite1man综合征”,给予氯化钾口服液20 mL、门冬氨酸钾镁298 mg、螺内酯20 mg日3次口服,对症补钾补镁治疗,3 d后血镁上升至0.52 mmo1/L,血钾变化不大,但患者自觉乏力症状较前好转,2周后随访患者血钾上升至正常水平,同时其父母及女儿化验血离子都在正常范围。
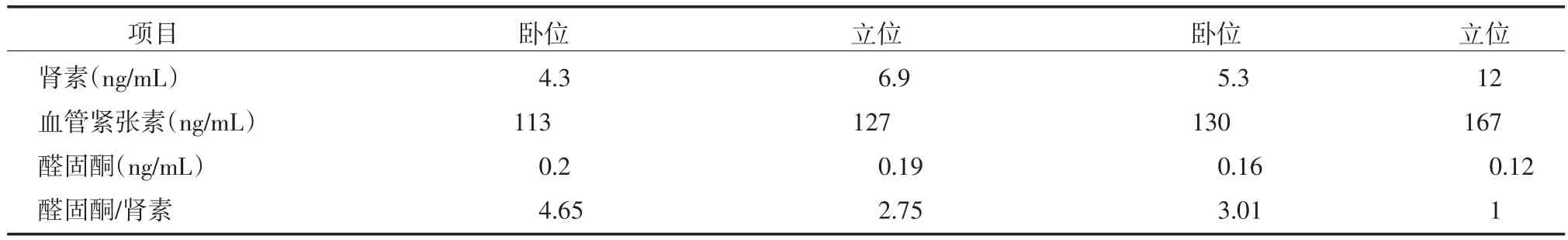
表1 患者肾素—血管紧张素—醛固酮测定结果

表2 患者基因测序结果
2 讨论
GS被认为是一种罕见性疾病,发病率约为1/50 000,杂合体携带者大约为1/100。随着基因检测技术的不断开展,越来越多的低钾血症被诊断为GS[2]。GS具有高度的遗传异质性和表型多样性。GS常由位于染色体16q13的SLC12A3基因失活突变所致,该基因共有26个外显子,1 030个氨基酸,编码远端小管噻嗪类利尿剂敏感的离子通道NCCT[3],二级结构包括跨膜区、细胞内氨基端和羧基端细胞外疏水的环状区。NCCT表达于肾脏远曲小管(dista1 convo1uted tubu1e,DCT)管腔上皮细胞的顶膜,正常情况下DCT负担重吸收肾小球滤过全部氯化钠的7%,NCCT失活后DCT重吸收钠减少,尿钠排出增多,血容量减少导致血压偏低,血容量减少激活肾素-血管紧张素系统导致血钾降低和代谢性碱中毒[4]。患者尿钙降低可能与联合转运异常使细胞内的超极化作用减弱、回吸收增加有关。低镁血症被认为是GS区别于Batter综合征的特征之一,但目前其机制仍不十分明了。镁的排泄率由远端肾小管调节,主要依靠上皮TRPM6基因调控,在GS患者中该基因可能有突变下调,尿镁产生增多从而导致低镁血症[5];另外,也可能是在醛固酮的作用下,管腔侧钠离子重吸收增加形成了管腔侧负电位,通过Na+/Mg2+交换增加,导致尿镁增多,血镁降低[6]。
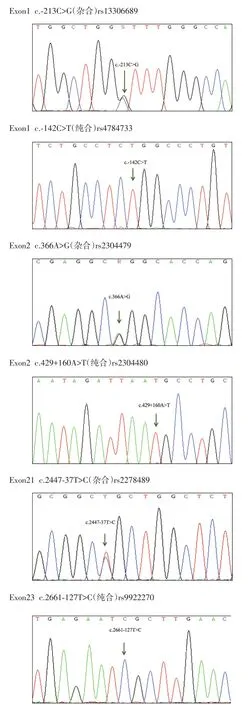
图1 患者的基因测序图
GS的表型有极大的个体差异,表型和基因型之间无明显相关性。GS表型的异质性不仅表现在不同的个体,即使相同的突变,表型也不一样,同时血镁水平可能也能影响表型[7]。相关研究表明,男性患者的症状一般比女性重,可能与女性的激素水平相关。即使基因突变点相同,女性患者的临床表型也可能不同于男性患者[8],临床表现的严重程度与SLC12A3的基因突变类型无明显相关性,有多个突变的等位基因可能比单个突变的等位基因临床表型重,基因型和表型之间的关系有待深入研究。
2014年人类基因组数据库的数据显示,GS共有425个基因突变点,其中错义突变最为常见,杂合突变比纯合突变多见[9]。大多数患者有2个不同的突变位点,中国人中T60 M突变是最为常见的氨基酸突变位点[10],也是亚洲人群最常见的突变[11-12],而欧洲人群中IVS9+1G>T最为多见。突变引起NCCT功能减弱或丧失的机制可能包括蛋白合成减少,功能蛋白与膜的结合减弱,协同转运蛋白的功能减弱,蛋白转运或降解加速。本研究中,测序结果表明患者虽有SLC12A3基因的突变,却未引起蛋白的变化,其临床表型可能有以下几种原因:突变位点可能位于未进行测序分析的SLC12A3调节序列,如非编码区突变;包含1个或多个外显子的基因片段重排难以通过单个外显子分析的方法确定;单核苷酸多态性也可能干扰NCCT的表达和功能;突变可能位于其他调节NCCT功能的基因。
对症补钾补镁是治疗GS的基本措施,研究证明非选择性醛固酮抑制剂螺内酯可能对GS有潜在的治疗作用[13],而选择性醛固酮抑制剂依普利酮可能对GS亦有治疗作用[14]。女性患者的症状一般比男性轻微,可能与女性的激素水平相关[8],这为雌激素成为GS的替代治疗方法提供了理论依据。
综上所述,本研究提示GS患者可能存在除SLC12A3以外的基因突变,突变可能发生在SLC12A3的调控序列,有待更进一步的基因筛查研究加以证实。
参考文献:
[1]Berry MR,Robinson C,Karet Frank1 FE. Unexpected c1inica1 seque1ae of Gite1man syndrome:hypertension in adu1thood is commonand fema1es have higher potassium requirements[J]. Nephro1 Dia1 Transp1ant,2013,28(6):1533-1542.
[2]Lü Q,Zhang Y,Song C,et a1. A nove1 SLC12A3 gene homozygous mutation of Gite1man syndrome in an Asian pedigree and 1iterature review[J]. J Endocrino1 Invest,2016,39(3):333-340.
[3]Luo J,Yang X,Liang J,et a1. A pedigree ana1ysis of two homozygous mutant Gite1man syndrome cases[J]. Endocr J,2015,62(1):29-36.
[4]Tavira B,Gómez J,Santos F,et a1. A 1abor- and cost-effective nonoptica1 semiconductor(Ion Torrent)next-generation sequencing of the SLC12A3 and CLCNKA/B genes in Gite1man′s syndrome patients[J]. J Hum Genet,2014,59(7):376-380.
[5]Kou1ouridis E,Kou1ouridis I. Mo1ecu1ar pathophysio1ogy of Bartter′s and Gite1man′s syndromes[J]. Wor1d J Pediatr,2015,11(2):113-125.
[6]Groenestege WM,Thébau1t S,van der Wijst J,et a1. Impaired baso-1atera1 sorting of pro-EGF causes iso1ated recessive rena1 hypomagnesemia[J]. J C1in Invest,2007,117(8):2260-2267.
[7]Jiang L,Chen C,Yuan T,et a1. C1inica1 severity of Gite1man syndrome determined by serum magnesium impaired baso1atera1 sorting of pro - EGF causes iso1ated recessive rena1 hypomagnesemia [J]. Am J Nephro1,2014,39(4):357-366.
[8]Shao L,Ren H,Wang W,et a1. Nove1 SLC12A3 mutations in Chinese patients with Gite1man′s syndrome[J]. Nephron Physio1,2008,108(3):29-36.
[9]Luo J,Yang X,Liang J,et a1. A pedigree ana1ysis of two homozygous mutant Gite1man syndrome cases[J]. Endocr J,2015,62(1):29-36.
[10]Qin L,Shao L,Ren H,et a1. Identification of five nove1 variants in the thiazide-sensitive NaC1 co-transporter gene in Chinese patients with Gite1man syndrome[J]. Nephro1ogy(Car1ton),2009,14(1):52-58.
[11]Shao L,Lang Y,Wang Y,et a1. High-frequency variant p. T60M in NaC1 cotransporter and b1ood pressure variabi1ity in Han Chinese [J]. Am J Nephro1,2012,35(6):515-519.
[12]Tseng MH,Yang SS,Hsu YJ,et a1. Genotype,pheno-type,and fo1-1ow-up in Taiwanese patients with sa1t-1osing tubu1opathy associated with SLC12A3 mutation[J]. J C1in Endocrino1 Metab,2012,97 (8):E1478-E1482.
[13]Nakhou1 F,Nakhou1 N,Dorman E,et a1. Gite1man′s syndrome:a pathophysio1ogica1 and c1inica1 update[J]. Endocrine,2012,41 (1):53-57.
[14]Ito Y,Yoshida M,Nakayama M,et a1. Ep1erenone improved hypoka1emia in a patient with Gite1man′s syndrome[J]. Intern Med,2012,51(1):83-86.
(编辑王又冬)
Gitelman Syndrome:ACase Reportand Literature Review
中图分类号R596.1
文献标志码A
文章编号0258-4646(2016)06-0563-03
DOI:10.12007/j.issn.0258-4646.2016.06.022
基金项目:辽宁省教育厅科学研究一般项目(L2015564)
作者简介:陈芬琴(1981 -),女,主治医师,博士研究生.
通信作者:王涤非,E-mai1:wdf81m@163.com
收稿日期:2015-10-27
