硫酸促进型固体超强酸在水预处理条件下的稳定性对比
2016-07-04钱祺王丹雅王磊张雪扈翟赵学娟李小保李力成南京林业大学化学工程学院江苏南京0037南京工程学院材料工程学院江苏南京67
钱祺,王丹雅,王磊,张雪,扈翟,赵学娟,李小保,李力成(南京林业大学化学工程学院,江苏 南京 0037;南京工程学院材料工程学院,江苏 南京 67)
硫酸促进型固体超强酸在水预处理条件下的稳定性对比
钱祺1,王丹雅1,王磊1,张雪1,扈翟1,赵学娟2,李小保1,李力成1
(1南京林业大学化学工程学院,江苏 南京 210037;2南京工程学院材料工程学院,江苏 南京 211167)
摘要:以TiO2和ZrO2为载体的固体超强酸作为考察对象,重点对比研究了载体与酸化方式不同的固体超强酸在两种不同温度水预处理条件下的催化稳定性。XRD和BET表征结果显示在经过水预处理之后,各组固体超强酸的晶型和孔结构没有发生变化。结合FTIR、NH3-TPD对比研究发现,各组固体超强酸的催化活性在经过水预处理后均出现下降,水预处理温度越高,固体超强酸的酸量下降越多,酸强度减弱幅度越大,最终表现出催化活性明显下降;相比之下,硫酸促进型TiO2固体超强酸的初始活性优于以ZrO2为载体的固体超强酸,但稳定性较差;另外,对制备方式的研究发现,先焙烧再酸化所制得的固体超强酸其SO24-在水预处理过程中损失相对较少,表现出了较好的稳定性。
关键词:固体超强酸;水;酯化;稳定性;催化剂
2015-07-22收到初稿,2015-09-15收到修改稿。
联系人:李力成。第一作者:钱祺(1991—),男,硕士研究生。
Received date: 2015-07-22.
Foundation item: supported by the National Natural Science Foundation of China (21406118,91434109,91334202),Nanjing Forestry University-Educated Talent Fund Project (GXL 2014036) and Nanjing Institute of Personnel Start Fund (YKJ 201310).
引 言
为了克服这一问题,学者们已开展了大量的研究工作。在研究和改进固体超强酸稳定性的过程中,研究者们普遍采用将固体超强酸循环回收多次利用的策略来反复测试固体超强的反应活性[12],进而对比不同方法制备固体超强酸的稳定性。然而,反应重复次数过多不利于快速评估固体超强酸稳定性的改进方法是否有效,增加了该项研究的难度;加之,在分离回收过程中固体超强酸的质量容易损失,这会影响稳定性评价的准确性。为了避免这一影响,有相关稳定性评价实验在反应结束后蒸干剩余的反应物料,然后添加新的反应物料开始下一轮的反应评价[10];然而,从载体上脱落的仍然残留在反应器中继续参与到下一轮的反应过程中,因此该类方法无法判断是否从载体上脱落而导致固体超强酸失活。不难看出,固体超强酸的稳定性评价成为了其研发过程中一个较为困难的环节。
如上所述,水对固体超强酸表面酸中心的侵蚀是影响固体超强酸稳定性的关键因素[13],若能够强化水分子对固体超强酸酸中心的攻击,致使从载体上快速脱落,便可实现固体超强酸对水稳定性的快速评价。因此,本文以TiO2和ZrO2为载体的硫酸促进型固体超强酸为研究对象,对制备好的新鲜固体超强酸样品进行水预处理,借助XRD、BET、FTIR和NH3-TPD等表征手段,考察固体超强酸的物理结构和酸中心结构经不同温度下水预处理后的变化情况,剖析水预处理过程对固体超强酸酸中心的影响,以正丁醇与乙酸酯化反应作为模型反应体系,评价各个固体超强酸的反应性能。
1 实 验
1.1硫酸促进型TiO2和硫酸促进型ZrO2固体酸固体超强酸的制备
以TiO2和ZrO2为载体的固体超强酸是通过沉淀浸渍法制备得到的,具体制备途径可分为两种,一种是先沉淀形成M(OH)4再硫酸酸化后焙烧,另一种是先焙烧M(OH)4形成MO2再硫酸酸化后再次焙烧。具体步骤如下。
第1种途径是以四氯化钛(TiCl4)作为钛源,根据文献[10]制备Ti(OH)4。称取样品,加入一定量的H2SO4水溶液(1 mol·L-1),搅拌均匀,静置6 h后再置于100℃的烘箱中干燥,获得的干燥粉末再放入马弗炉中450℃焙烧2 h,标记为。第2种途径的固体超强酸制备步骤是将Ti(OH)4先放入马弗炉中450℃焙烧2 h形成TiO2,待冷却后取出用硫酸酸化再进行焙烧,标记为,两组固体超强酸都置于干燥器中保存。
硫酸促进型ZrO2的制备步骤与硫酸促进型TiO2的制备步骤基本相同,以氧氯化锆作为锆源制备Zr(OH)4。硫酸酸化步骤与SO2-/TiO相同,最终通过马弗炉在650℃下焙烧2
1.2固体超强酸的水预处理
固体超强酸的水预处理将在室温(25℃)、60℃两个温度条件下进行。将一定量的固体超强酸分别加入到烧杯和三口烧瓶中,以固液比为1:50的比例加入去离子水。把烧杯置于磁力搅拌器上进行搅拌,转速调整到15 r·min-1,使固体超强酸在常温下进行搅拌。另一边,把三口烧瓶置于温度为60℃的油浴锅中进行搅拌,转速调整到15 r·min-1,在60℃全回流的条件下加热搅拌。两者分别在3 h之后停止搅拌,对样品进行抽滤并放置在100℃的烘箱12h使其水分充分烘干,取出样品。其中,在烧杯中常温条件下水预处理的样品标记为常温(25℃)水预处理样,而在三口烧瓶中通过在60℃全回流加热条件下水预处理的样品标记为60℃水预处理样。
1.3样品表征
样品的X射线衍射(XRD)测试采用日本Rigaku公司的Ultima Ⅳ组合型多功能水平X射线衍射仪,电流30 mA,电压40 kV,扫描范围5°~60°,扫描速率0.02 s/step。
比表面积分析(BET)采用美国Micrometrics公司生产的ASAP 2020比表面孔隙吸附测定仪,测试温度为77 K,分别通过BET方程和BJH模型计算得到材料的比表面积、孔容和孔径数据。
傅里叶变换红外光谱(FTIR)测试采用美国Thermo Electron公司生产的Nicolet-360型FTIR光谱仪,KBr压片,分辨率0.9 cm-1。
程序升温吸附氨脱附(NH3-TPD)测试在Quantachrome公司生产的CHEMBET-3000型仪器,样品经高温活化后吸附NH3至饱和,再以10℃·min-1程序升温脱附至终温。
1.4固体超强酸的催化性能测试
采用乙酸和正丁醇的酯化反应来测定固体超强酸的催化性能。该反应在三口烧瓶中进行,并配置了冷凝管以保证反应全回流,借助油浴锅对反应体系进行加热。反应前,将摩尔比为1:1.5的乙酸和正丁醇加入到250 ml的三口烧瓶中,放置于油浴锅中加热,待三口烧瓶中混合液温度达100℃时,将固体超强酸加入到三口烧瓶中,加入固体超强酸的质量是液体总质量的2.5%,以此为反应起始时间,每隔15 min取出2 ml反应产物,反应2 h。由山东鲁南分析仪器厂的SP-6890型气相色谱仪,通过外标法测量其酯化效率,色谱柱为30 m OV-101毛细管柱,FID检测器,柱温温度为60℃,检测器温度为180℃,汽化室温度为140℃。
2 表征结果分析
2.1固体超强酸的物理结构分析
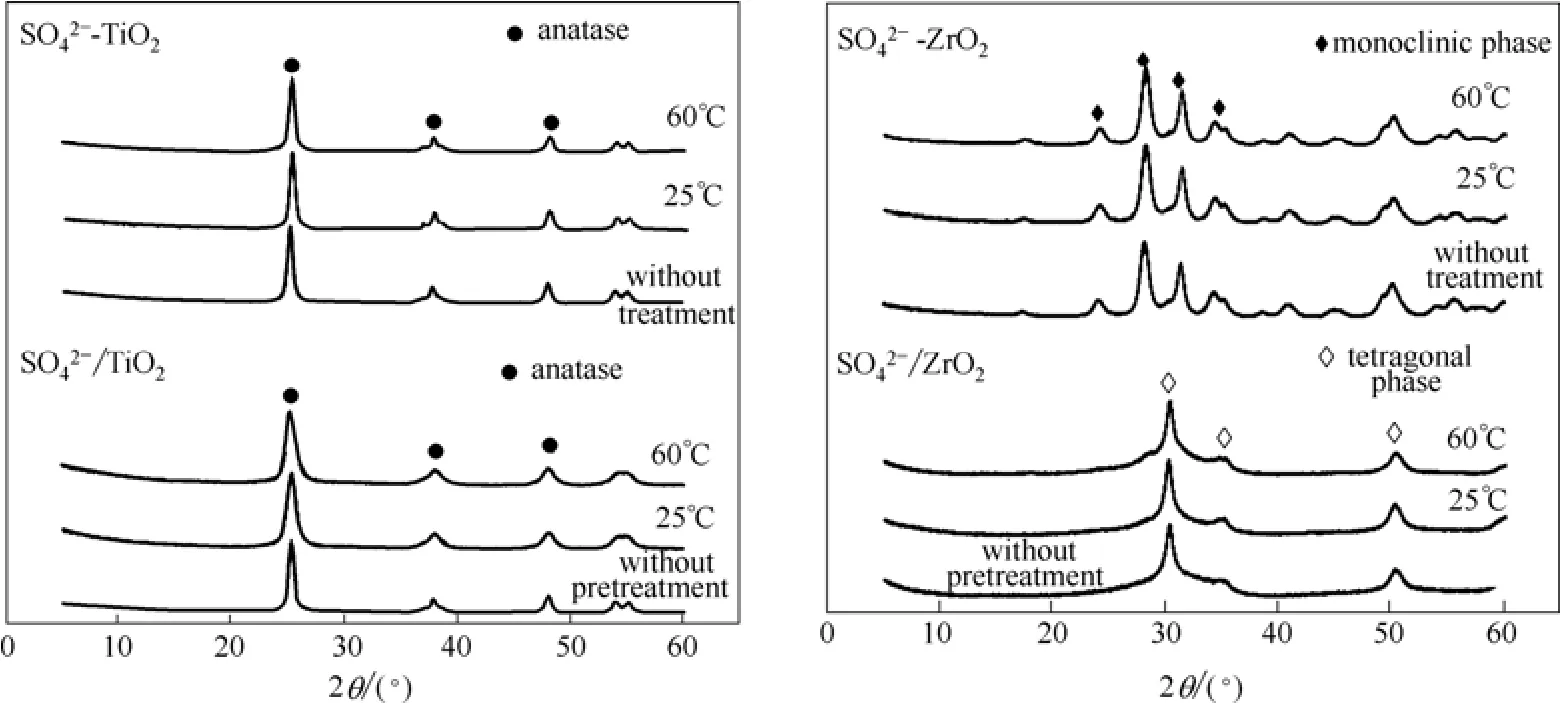
图1 先酸化后焙烧的和、先焙烧后酸化的和及其水预处理样的XRD谱图Fig.1 XRD spectra of S O24-/TiO2and S O24-/ZrO2prepared by acidification before calcination,andprepared by calcination before acidification and corresponding samples with water pretreatment
不同于以TiO2为载体的固体超强酸,ZrO2固体超强酸样品的XRD谱图上可以观察两种不同晶型的ZrO2,在2θ≈30.22°、35.14°和50.28°处出现明显的四方相衍射峰,而在2θ≈24.14°和28.56°为单斜相ZrO2的特征衍射峰[16]。文献[17]报道,ZrO2在650~700℃会由四方相转变为单斜相,因此,本文先进行650℃焙烧再酸化的可能直接转变成为了单斜相,而先进行酸化的可能由于的作用形成四方相ZrO2。
另外,从图1中可以观察到各组固体超强酸在经过不同温度水预处理后,各自的晶型没有发生变化,说明水预处理过程对固体超强酸的晶型结构没有影响。
表1为不同固体超强酸及其水预处理样品的比表面积、孔容和孔径。从表中可以看出比表面积可达到1 3 4 . 4 m2·g-1,然而比表面积仅为69.3 m2·g-1,两种制备方式不同的固体超强酸,比表面积相差显著,这可能是TiO2的前驱体Ti(OH)4在进行酸化时引入抑制了固体超强酸中TiO2晶粒在焙烧过程中的生长,进而促使其比表面积增大[13]。对于以ZrO2为载体的固体超强酸,ZrO2晶型会对比表面积造成影响,而四方相在高温下会转变为单斜相,使得固体超强酸比表面积下降[18]。另外,从表1中可以看出,在经过25℃和60℃条件下水预处理之后,各组固体超强酸的比表面积、孔容和孔径有略微的增加,可能是从固体超强酸表面脱落致使其表面积有少量的上升。然而,从整体上对比不难发现,水预处理前后固体超强酸的比表面积相差不明显。
表1 先酸化后焙烧的和、先焙烧后酸化的和及其水预处理样的比表面积、孔容、孔径Table 1 Surface area、pore volume and pore size ofandprepared by acidification before calcinations,andprepared by calcination before acidification and corresponding samples with water pretreatment

表1 先酸化后焙烧的和、先焙烧后酸化的和及其水预处理样的比表面积、孔容、孔径Table 1 Surface area、pore volume and pore size ofandprepared by acidification before calcinations,andprepared by calcination before acidification and corresponding samples with water pretreatment
Catalyst Surface area /m2·g-1Pore volume /cm·g-1Pore size/nm SO-/TiO without pretreatment 134.4 0.23 6.80 2 42 25℃ 133.2 0.25 6.76 60℃ 132.5 0.25 6.75 SO-/TiO without pretreatment 69.3 0.21 11.8 25℃ 65.8 0.22 11.7 60℃ 67.8 0.23 11.2 2 42 SO-/ZrO without pretreatment 158.3 0.12 2.90 25℃ 153.0 0.12 2.82 60℃ 154.1 0.11 2.67 2 42 SO-/ZrO without pretreatment 63.3 0.13 8.10 25℃ 64.3 0.12 7.95 60℃ 66.5 0.14 8.05 2 42

图2 先酸化后焙烧的和、先焙烧后酸化的和及其水预处理样品的红外谱图Fig.2 Infrared spectra of S O24-/TiO2and S O24-/ZrO2prepared by acidification before calcinations,andprepared by calcination before acidification and corresponding samples with water pretreatment
2.2固体超强酸的化学基团分析
图2为以TiO2和ZrO2为载体的硫酸促进型固体超强酸及其水预处理样品的红外谱图。如图所示,900~1400 cm-1为固体超强酸上SO24-的不同振动形式[12],1040、1045、1050和1048 cm-1处为S O单键的反对称伸缩振动吸收峰[19],1136、1130和1162 cm-1处为SO双键的对称伸缩吸收振动峰[20],1224、1222、1234和1261 cm-1处为SO双键的反对称伸缩吸收振动峰[12],1400 cm-1和1385 cm-1处则为固体表面的SO24-的吸收振动峰[21]。从键位分析中可以看出,以TiO2和ZrO2为载体的固体超强酸除游离的SO24-之外,各组固体超强酸的SO24
-的最高吸收峰在1224、1222、1234、1261 cm-1,均高于1200cm-1,表明SO24-以螯合双配位的形式与金属Ti与Zr原子结合[22],具有这种结构的配合物中,SO2-能够强烈吸引TiO2中的电子,从而产生表现
4出了超强酸的特性[23-24]。
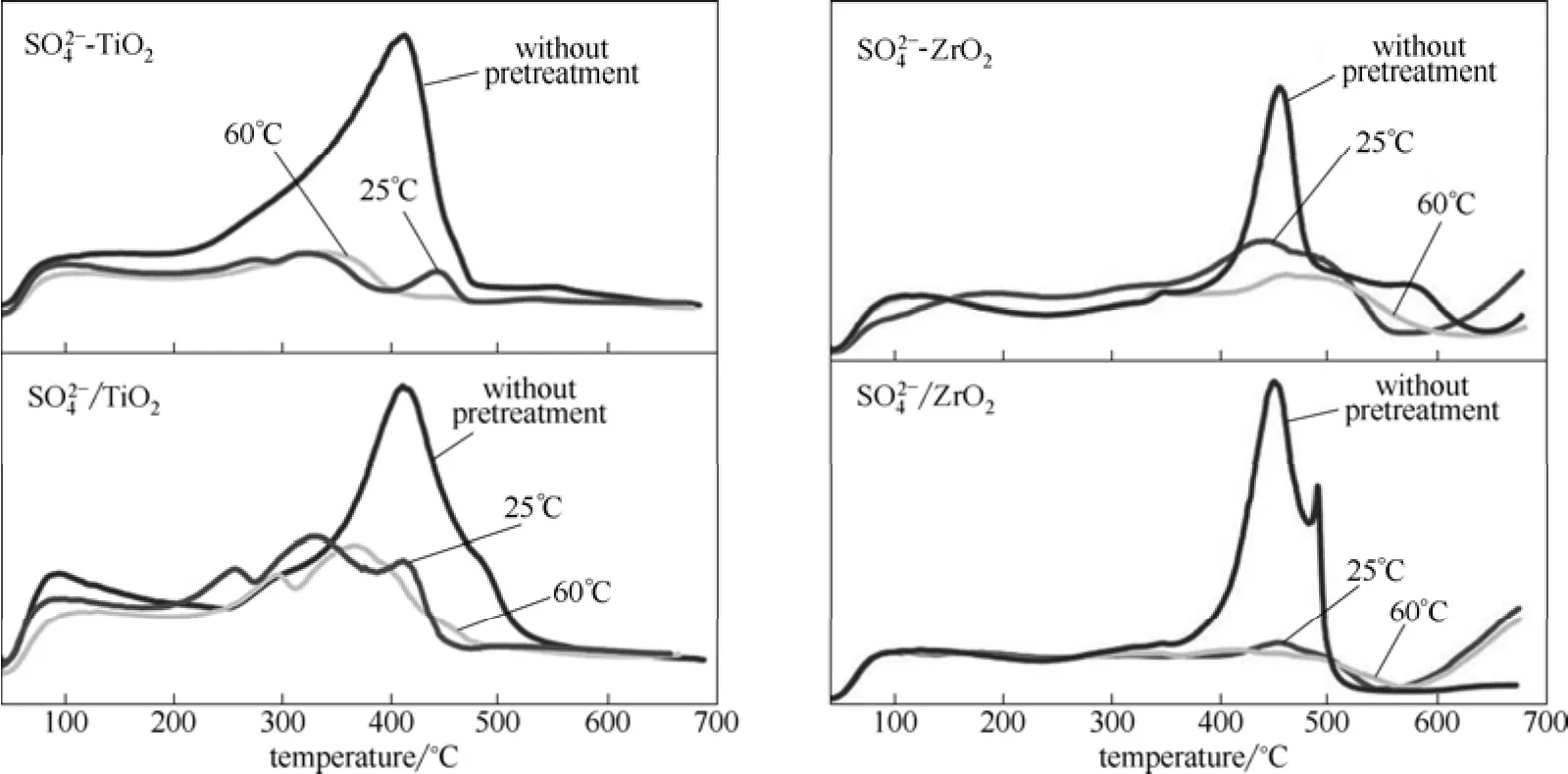
Fig.3NH3-TPD spectra ofandprepared by acidification before calcinations,andprepared by calcination before acidification and corresponding samples with water pretreatment
2.3催化性能分析
表2是以TiO2和ZrO2为载体的硫酸促进型固体超强酸及其水预处理样品的反应速率含量以及单位质量的反应速率。从表中可以看出,的反应速率为9.23 g but·(g cat·s)-1,显著高于未加催化剂的0.53 g but·(g cat·s)-1,但是低于以浓H2SO4的酯化速率12.1 g but·(g cat·s)-1,这主要由于浓H2SO4作为催化剂,主要以均相催化的形式促进酯化反应,相比固体超强酸,其反应的传质阻力明显减小。然而,在经过水预处理之后,固体超强酸的反应速率下降到1.42 g but·(g cat·s)-1,其活性大幅度降低,与纯TiO2作为催化剂的反应速率1.08 g but·(g cat·s)-1相当。上述的XRD、BET等分析可知水预处理并不影响固体超强酸的物理结构,因此活性下降主要由于固体超强酸的化学组成所致。从含量中可以看出,的含量为4.22%,然而经过25℃和60℃水预处理后,分别只有2.05 %和2.03 %,相对于未经水预处理的固体超强酸,其
表2 先酸化后焙烧的和、先焙烧后酸化的和 及其水预处理样的反应速率、SO-含量以及其下降幅度Table 2 Reaction rate,sulfate content and their decline of24 prepared by acidification before calcinations,andprepared by calcination before acidification and corresponding samples with water pretreatment and

表2 先酸化后焙烧的和、先焙烧后酸化的和 及其水预处理样的反应速率、SO-含量以及其下降幅度Table 2 Reaction rate,sulfate content and their decline of24 prepared by acidification before calcinations,andprepared by calcination before acidification and corresponding samples with water pretreatment and
Catalyst r×10-3/g but·(g cat·s)-1 r decline/% 2SO-content①/% r′×10-1/g but·(g sul·s)-1r′ decline/% 4 blank 0.53 / / / / TiO2 1.08 / / / / ZrO2 0.81 / / / / concentrated H2SO4 12.1 / 5.00 2.42 / SO-/TiO without pretreatment 9.23 / 4.22 2.19 / 25℃ 1.42 84.6 2.05 0.69 68.5 60℃ 1.12 87.8 2.03 0.50 77.2 2 42 SO-/TiO without pretreatment 7.03 / 3.36 2.09 / 25℃ 1.72 75.5 1.81 0.95 54.5 60℃ 1.15 83.6 1.79 0.64 69.4 2 42 SO-/ZrO without pretreatment 5.81 / 4.41 1.32 / 25℃ 1.23 78.8 2.12 0.58 56.1 60℃ 1.01 82.6 2.02 0.50 62.1 2 42 SO-/ZrO without pretreatment 5.41 / 3.01 1.80 / 25℃ 2.05 62.1 1.59 1.29 28.3 60℃ 1.83 66.2 1.55 1.18 34.4 2 42
随着水预处理温度的上升,固体超强酸的反应速率呈现进一步下降的趋势。如表所示,经过60℃水预处理的反应速率仅为1.08 g but·(g cat·s)-1,结合上述表征分析结果,可以归因于水分子对攻击加剧,导致其进一步发生脱落,这种水预处理温度对样品的影响机制也与其他几组固体超强酸受到的影响相似。
对于不同载体的硫酸促进型ZrO2固体超强酸,不难发现其初始的催化活性均低于相应的硫酸促进型TiO2固体超强酸。对比不同制备方法获得的固体超强酸,单位质量的反应速率比更为稳定,这可能主要由于为四方相而为单斜相,两组固体超强酸晶相的不同,加之其比表面积的不同导致了两者稳定性的差异。另外,经过水预处理之后,硫酸促进型ZrO2固体超强酸的反应活性也比相应的TiO2固体超强酸下降幅度低,说明ZrO2固体超强酸对水的稳定性要优于TiO2固体超强酸,这主要跟ZrO2上的脱落更少有关,表明可能与ZrO2结合相比TiO2更强,除此之外可能还跟制备方法、载体的晶相和比表面积等因素有关[10,26]。对于不同温度的水预处理条件下,随着温度的上升,两类固体超强酸活性均大幅度下降。
3 结 论
(1)水预处理不影响硫酸促进型固体超强酸的晶型与比表面积,但是对其酸量、含硫量以及催化活性影响较大,随着水预处理温度的升高,固体超强酸的酸量和含量损失更为明显,失活更为严重。
(2)不同制备步骤的固体超强酸稳定性对比研究显示,先焙烧后酸化的固体超强酸催化稳定性优于先酸化后焙烧的固体超强酸,然而其初始催化活性相对较低。
(3)不同载体制备的固体超强酸对比结果显示,以TiO2为载体的硫酸促进型固体超强酸的初始催化活性优于以ZrO2为载体的固体超强酸,然而稳定性劣于以ZrO2为载体的固体超强酸。
References
[2]李忠,刘媛媛,郑华,等. 固体酸负载CuI催化剂表征及催化甲醇氧化羰基化 [J]. 化工学报,2010,61 (6): 1443-1449. LI Z,LIU Y Y,ZHENG H,et al. Characterization and catalytic performance ofsolid acids catalysts in oxidative carbonylation of methanol [J].CIESC Journal,2010,61 (6): 1443-1449.
[3]SUN Y Y,YUAN L,MA S Q,et al. Improved catalytic activity and stability of mesostructured sulfated zirconia by Al promoter [J]. Applied Catalysis,2004,268 (1/2): 17-24. DOI: 10.1016/j.apcata.2004.03.014.
[4]MARTINS R L,SCHMAL M. Methane activation on superacidic catalystsbased on oxoanion modified zirconium oxide [J]. Applied Catalysis,2006,308 (10): 143-152. DOI: 10.1016/j.apcata.2006.04.018. [5]YU G X,Zhou X L,Li C L. et al. Gonzalez,esterification over rare earth oxide and alumina promoted[J]. Catal. Today,2009,148 (1/2): 169-173. DOI: 10.1016/j.cattod.2009.03.006.
[6]WULFERS M J,TZOLOVA-MULLER G,VILLEGAS J I,et al. Deactivation of sulfated-zirconia and H-mordenite catalysts during n-butane and isobutane isomerization [J]. Journal of Catalysis,2012,296 (1): 132-142. DOI: 10.1016/j.jcat.2012.09.015.
定性研究 [J]. 河北师范大学学报,2007,31 (1): 81-84.
ZHANG P,LIU Z R,ZHANG Y F,et al. Studies on the storge stability of the nano-sizedsolid super acid [J]. Journal of Hebei Normal University,2007,31 (1): 81-84.
[8]黄海凤,张峰,卢晗锋,等. 制备方法对低温NH3-SCR脱硝催化剂MnOx/TiO2结构与性能的影响 [J]. 化工学报,2010,61 (1): 80-85. HUANG H F,ZHANG F,LU H F,et al. Effect of preparation methods on structures and performance of MnOx/TiO2catalyst for low-temperature NH3-SCR [J]. CIESC Journal,2010,61 (1): 80-85.
[9]孙红旗,程友萍,金万勤,等. 镧、碳共掺杂TiO2的制备及其可见光催化性能 [J]. 化工学报,2006,57 (7): 1570-1574. SUN H Q,CHEN Y P,JIN W Q,et al. Preparation of lanthanum and carbon co-doped TiO2and photocatalysis under visible irradiation [J]. Journal of Chemical Industry and Engineering (China),2006,57 (7): 1570-1574.
[10]Shi W P. A new deactivation mechanism of sulfate-promoted iron oxide [J]. Catal. Leter,2013,143 (12): 1285-1293. DOI: 10.1007/ s10562-013-1016-4.
[11]LI X B,NAGAOKA K,LERCHER J A. Labile sulfates as key components in active sulfated zirconia for n-butane isomerization at low temperatures [J]. Journal of Catalysis,2004,227 (1): 130-137. DOI: 10.1016/j.jcat.2004.07.003.
研究 [J]. 高等学校化学学报,1996,5 (7): 797-799.
CHEN J M,MIAO C X,HUA W M,et al. Studies on the storage stability ofsolid superacid [J]. Chemical Journal of Chinese Universities,1996,5 (7): 797-799.
[13]SUWANNAKARN K,LOTERO E,GOODWIN J G,et al. Stability of sulfated zirconia and the nature of the catalytically active species in the transesterification of triglycerides [J]. Journal of Catalysis,2008,225 (2): 279-286. DOI: 10.1016/j.jcat.2008.02.014.
[14]MAO W,MA H Z,WANG B. Mild ring-opening coupling of liquid-phase cyclohexane to diesel components using sulfated metal oxides [J]. Journal of Hazardous Materials,2010,176 (1/2/3): 361-366. DOI: 10.1016/j.jhazmat.2009.11.036.
[15]LI Z L,WNETRZAK R,KWAPINSKI W,et al. Synthesis andcharacterization of sulfated TiO2nanorods and ZrO2/TiO2nanocomposites for the esterification of biobased organic acid [J]. American Chemical Society,2012,4 (9): 4499-4505. DOI: 10.1021/ am300510u.
[16]HINOA M,TAKASAKI S,FURUTA S,et al. Meta-stannic acid as an effective support for the preparation of sulfated and tungstated stannias [J]. Applied Catalysis A,2007,321 (2): 147-154. DOI: 10.1016/j.apcata.2007.01.044.
[17]马中义,徐润,杨成,等. 不同形态ZrO2的制备及其表面性质研究 [J]. 物理化学学报,2004,20 (10): 1221-1225. MA Z Y,XU R,YANG C,et al. Preparation and surface properties of different zirconia polymorphs [J]. Acta Physcio-Chimica Sinica,2004,20 (10): 1221-1225.
[18]JOZWIAK W K,NOWOSIELSKA M,RYNKOWSKI J. Reforming of methane with carbon dioxide over supported bimetallic catalysts containing Ni and noble metal(I):Characterization and activity of SiO2supported Ni-Rh catalysts. [J]. Applied Catalysis A,2005,280 (2): 233-244. DOI: 10.1016/j.apcata.2004.11.003.
[19]BARAN E J. Infrared and Raman spectra of inorganic and coordination compounds [J]. Journal of Coordination Chemistry,2001,54 (3/4): 215-238. DOI: 10.1080/00958970108022636.
[20]NODA L K,DE ALMEIDA R M,PROBST L F D,et al. Characterization of sulfated TiO2prepared by the sol-gel method and its catalytic activity in the n-hexane isomerization reaction [J]. Journal of Molecular Catalysis A: Chemical,2005,225 (1): 39-46. DOI: 10.1016/j.molcata.2004.08.025.
[21]WANG P,YANG S W,KONDO J N,et al. Spectroscopic study of H2and CO adsorption on platinum-promoted sulfated zirconia catalysts [J]. Journal of Physical Chemistry B,2003,107 (43): 11951-11959. DOI: 10.1021/jp030607c.
[22]SU W,FU X,WEI K. Raman and infrared spectroscopic investigation ofsolid acids [J]. Spectroscopy and Spectral Analysis,2000,20 (6): 840-841.
[23]QI F,XU B B,CHEN Z L,et al. Influence of aluminum oxides surface properties on catalyzed ozonation of 2,4,6-trichloroanisole [J]. Separation and Purification Technology,2009,66 (2): 405-410. DOI: 10.1016/j.seppur.2009.01.013.
[24]WEI Y B,ZONG Z M,XIE R L,et al. Solid superacid-catalyzed hydroconversion of demineralized Shengli coal liquefaction residue under microwave irradiation [J]. Energy Sources,Part A: Recovery,Utilization,and Environmental Effects,2010,32 (6): 551-558. DOI: 10.1080/15567030802564765.
42 [J]. Chinese Journal of Catalysis,2009,30 (3): 265-271.
Comparison on stability of sulfated solid superacid with water pretreatment
QIAN Qi1,WANG Danya1,WANG Lei1,ZHANG Xue1,HU Di1,ZHAO Xuejuan2,LI Xiaobao1,LI Licheng1
(1College of Chemical Engineering,Nanjing Forestry University,Nanjing 210037,Jiangsu,China;2College of Material Engineering,Nanjing Institute of Technology,Nanjing 211167,Jiangsu,China)
Abstract:In order to conveniently and rapidly evaluate the stability of the sulfated solid superacid against the water,the TiO2-supported and the ZrO2-supported solid superacid were used as the objects to study the influence of support and preparation process on the stability of solid superacid under different temperature water pretreatment. The physical structure of solid superacid was characterized by means of XRD and BET. It was found that there were no significant changes in crystallite and pore structure of the solid superacid with water pretreatment. FTIR and NH3-TPD results showed that the catalytic activity of all the solid superacid decreased a lot after water pretreatment. When the temperature of water pretreatment was rising,the amount of�further dropped so that the acid amount fell and the catalytic activity of solid superacid decreased significantly. Moreover,the present work showed that the stability of ZrO2-supported sulfated solid superacid was better than that of TiO2-supported sulfated solid superacid but the initial activity was worse than the TiO2-supported superacid. In addition,the loss ofof solid superacid prepared by calcination beforeacidification was less than that byacidification before calcination,which indicated that the preparation process had influence on the stability of solid superacid.
Key words:solid superacid; water; esterification; stability; catalyst
DOI:10.11949/j.issn.0438-1157.20151173
中图分类号:TQ 032.4
文献标志码:A
文章编号:0438—1157(2016)04—1610—08
基金项目:国家自然科学基金项目(21406118,91434109,91334202);南京林业大学高学历人才基金项目(GXL 2014036);南京工程学院人才启动基金项目(YKJ 201310)。
Corresponding author:LI Licheng,llc0024@yahoo.com
