正庚烷脱氢生成烯烃反应机理的模拟研究
2016-07-01马爱增代振宇
于 宁, 龙 军, 周 涵, 马爱增, 代振宇
(中国石化 石油化工科学研究院, 北京 100083)
正庚烷脱氢生成烯烃反应机理的模拟研究
于宁, 龙军, 周涵, 马爱增, 代振宇
(中国石化 石油化工科学研究院, 北京 100083)
摘要:采用基于密度泛函理论的量子化学方法研究了催化重整过程中正庚烷脱氢生成烯烃的反应过程。通过对比2条不同的反应路径得出, Pt原子在脱氢反应中生成的Pt-H活性中心具有吸取单电子的能力,具有较强的脱除氢自由基的催化能力。反应过程中,正庚烷首先在0价态的Pt原子表面发生化学吸附,随后发生脱除氢自由基反应,生成庚基自由基和Pt-H活性中心,优先生成2-庚基自由基,最低反应能垒为75.89 kJ/mol;庚基自由基直接与Pt-H催化剂活性中心发生化学吸附,进一步发生脱除氢自由基反应,生成庚烯与Pt-H2,优先生成2-庚烯,最低反应能垒为17.52 kJ/mol;最终,庚烯从Pt-H2表面发生脱附,随后Pt-H2发生脱附反应生成H2和再生的0价态的单Pt催化剂。该反应路径中最大反应能垒为75.89 kJ/mol。实验证明,正庚烷脱氢生成正庚烯的反应过程中优先生成2-庚烯。
关键词:正庚烷; 烷烃脱氢; Pt; 反应机理; 分子模拟
催化重整的主反应为六元环烷烃脱氢生成芳烃、烷基环戊烷脱氢异构生成芳烃和链烷烃脱氢环化生成芳烃的反应。其中,链烷烃脱氢环化生成芳烃的反应最难发生,提高链烷烃脱氢环化的转化率成为国内外研究的热点[1]。
正庚烷常被作为考察链烷烃脱氢环化反应的模型化合物[2-4]。Mills等[5-6]提出,在含Pt的双功能催化剂作用下,正庚烷首先在Pt金属表面脱除氢自由基生成烯烃;随后,烯烃作为重要的中间产物,发生芳构化等反应。前人对烷烃脱氢生成烯烃的反应进行了大量的实验研究[7-9],并得出烷烃脱氢生成烯烃反应的平衡常数较小,平衡转化率较低;平衡产物中,1-烯烃含量极少,绝大部分是内烯烃。
笔者等[10]曾采用分子模拟方法研究正庚烷脱氢环化反应,得出在无催化剂作用下,正庚烷分子的仲碳C—H键优先发生均裂生成烷基自由基和氢自由基,均裂能在433.80~434.83 kJ/mol之间;中间产物烷基自由基中,与自由基碳相邻的仲碳位的C—H键容易发生均裂生成烯烃和氢自由基,均裂能在187.11~209.18 kJ/mol范围;反应产物主要为2-庚烯、3-庚烯。在单原子Pt催化剂作用下,正庚烷均裂仲碳位C—H键的反应能垒在75.90~78.51 kJ/mol范围,中间产物烷基自由基中与自由基碳相邻的仲碳位的C—H键均裂的反应能垒为99.63 kJ/mol。
本研究中,笔者采用基于密度泛函理论的量子化学方法,从Pt和正庚烷二者在反应过程中的轨道排布以及化学键变迁等角度,深入探讨了正庚烷脱氢生成庚烯的反应机理。
1正庚烷脱氢生成庚烯反应的模拟方法和思路
采用Accelrys公司的分子模拟软件Materials Studio 5.5构建不同化合物的结构模型。采用Adsorption Locator模块研究正庚烷在催化剂表面的吸附构型。利用基于密度泛函理论的量子力学从头计算模块DMol3优化结构,并计算体系的自由能,从而获得C—H键均裂反应的反应能垒;对比不同反应路径中基元反应的反应能垒,确定最优反应路径。在DMol3计算中,选用泛函为GGA-PW91,收敛精度为能量0.05 kJ/mol、受力10.5×1013N/mol、位移5×10-13m。
在计算过程中,首先对Pt原子的外层价电子轨道和反应物在催化剂表面的吸附状态进行分析,随后对比研究了正庚烷在Pt催化剂表面的2种不同反应路径,最终获得了正庚烷在Pt催化剂上脱氢生成正庚烯的最佳反应路径。同时,通过实验手段对模拟获得的部分结果进行验证。
2实验部分
2.1原料及催化剂
正庚烷,分析纯,购买自百利威试剂公司。双功能催化剂Pt-γ-Al2O3-Cl,由中国石化石油化工科学研究院提供,其中,w(Pt)=0.3%,w(Cl)=1.0%。
2.2催化剂评价及产物组成分析方法
采用Meryer公司08141型微反-色谱系统评价催化剂反应性能。催化剂装填量1 mL、温度370~500℃、压力700 kPa、LHSV 2.0 h-1,氢/油体积比1000。
按照石油化工科学研究院分析方法GC12-DHA3分析液相产物单体烃PONA组成,按照石油化工科学研究院分析方法GC43-OTHER进行杂项分析。
3结果与讨论
3.1Pt原子催化原理分析
Cs-HRSTEM表征结果得出,Pt-γ-Al2O3催化剂中Pt大多数以单原子状态分散;TPR实验表明Pt-γ-Al2O3催化剂中Pt在高氢分压的模拟使用状态时为0价态[11]。分析得出,在Pt-γ-Al2O3催化剂中载体对Pt原子的电子价态影响极小。为了适当减少模型计算量,在本研究的模拟计算过程中忽略载体对Pt的电子价态影响,并采用单个Pt原子作为重整催化剂中的金属活性中心。
0价态Pt原子的最外层价电子轨道排布为5d96s1,如图1所示。从图1可见,Pt原子的5d电子轨道与6s电子轨道中均存在1个未配对的自由电子,处于电子未饱和状态,从而具有吸取2个电子的能力,使5d电子轨道与6s电子轨道达到全满状态。
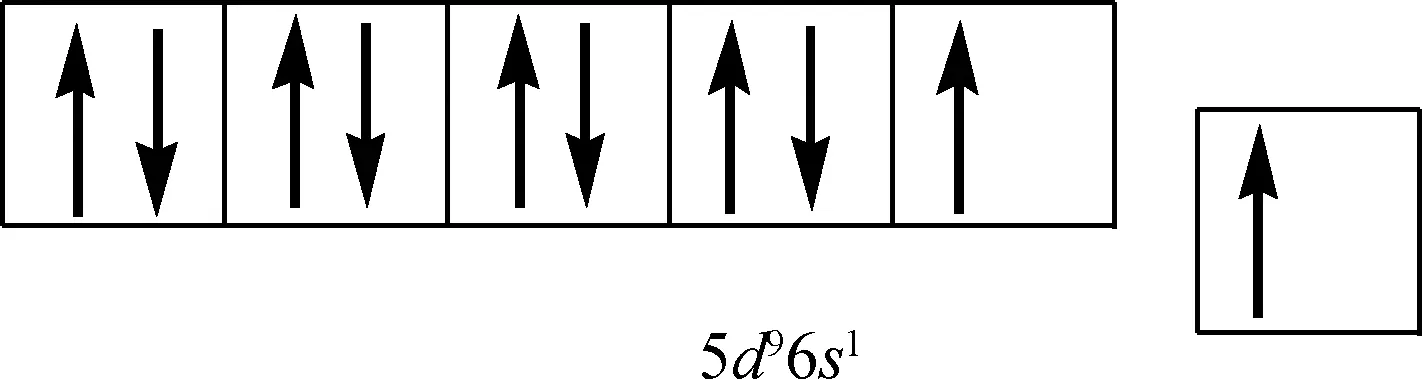
图1 Pt原子的最外层电子结构示意图
在烷烃脱氢反应中,Pt原子作为催化活性中心可以从烷烃中的C—H键中夺取含有单电子的氢自由基。
3.2正庚烷在催化剂表面的吸附过程
参考彭红建[12]、Jerzy等[13]的建模方法,选取γ-Al2O3载体中比表面积比例最大的(440)晶面。采用Visualizer模块切出γ-Al2O3晶胞中的(440)晶面,构建1个原子数为16×16、6层原子厚度的模型作为理想状态下的催化剂表面。由于催化剂表面Pt负载量为千分之一级单位,催化剂表面单位面积上的Pt含量极低,因此吸附模拟研究采用的简化表面模型中没有添加单个Pt原子。
采用Adsorption Locator模块模拟研究了正庚烷在催化剂表面的最稳定吸附结构,其构象如图2所示。由俯视图(a)和侧视图(b) 2种视角看出,正庚烷分子结构中外围的H原子与催化剂表面优先接触,从而可进一步发生脱氢反应。相反,分子结构中的C原子与催化剂表面无法接触,正庚烷很难在平整的载体表面发生C—C键的断裂反应。因此,从吸附角度说明了涉及C—H键断裂的结构不敏感反应(如脱氢反应)容易在平整的催化剂表面发生,而涉及C—C键的结构敏感反应[14-16](如烷烃的氢解反应)很难在平整的催化剂表面发生。经测量正庚烷分子结构中的H原子与催化剂表面的吸附距离为0.216 nm。
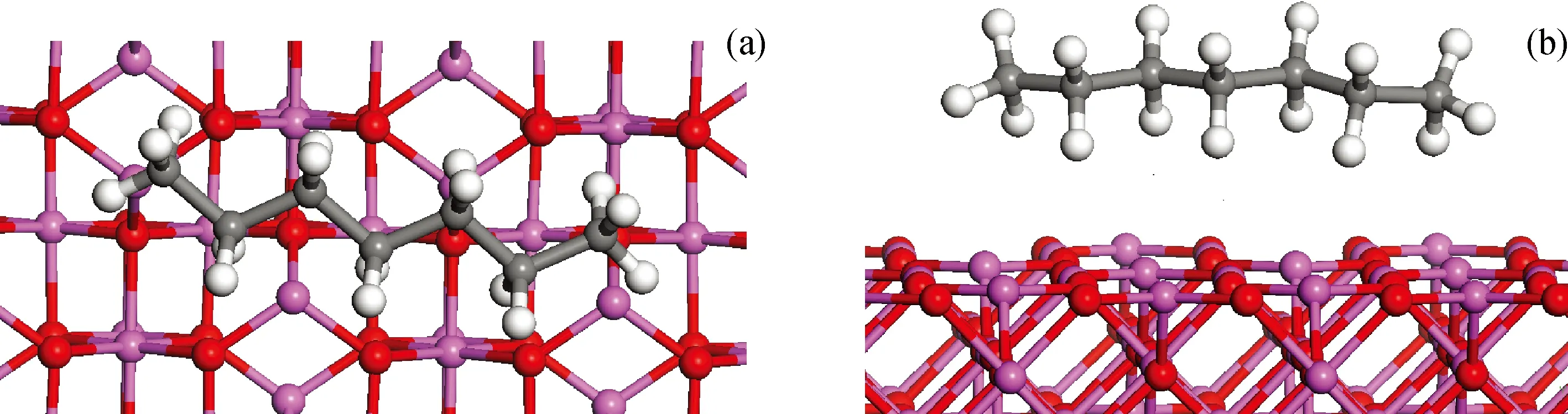
图2 正庚烷在L酸表面吸附构象
3.3Pt催化剂对正庚烷脱氢生成烯烃的催化过程
在无催化剂作用下,正庚烷脱氢生成烯烃的反应遵循自由基反应机理,分两步进行[10]。正庚烷分子的仲碳C—H键优先发生均裂生成烷基自由基和氢自由基,烷基自由基中,与自由基碳相邻的仲碳位的C—H键容易发生均裂生成烯烃,主要为2-庚烯、3-庚烯。
与无催化剂作用相比,在单原子Pt催化剂作用下的实际催化反应中,正庚烷分子在2次脱除氢自由基时的化学键断裂顺序没有改变;但是受Pt催化剂作用,化学键断裂的反应能垒降低。
由于Pt原子外层电子轨道存在2个未饱和的电子轨道,当0价态的单Pt原子吸附1个氢自由基生成Pt-H催化活性中心后,其中的Pt原子仍存在1个未饱和的单电子轨道,该轨道具有吸附电子的作用,因此可再吸附1个氢自由基,因此,正庚烷在Pt催化剂表面脱氢生成正庚烯的过程存在2条反应路径。
路径一:正庚烷在0价态的单Pt催化剂作用下,脱除氢自由基生成庚基自由基;庚基自由基脱附、扩散、吸附在另一个0价态的单Pt催化剂表面;庚基自由基在0价态的单Pt催化剂上脱除氢自由基,生成庚烯和氢自由基;庚烯从催化剂脱附,而氢自由基通过氢溢流方式合并生成氢气并脱附。该反应路径中的脱氢反应能垒计算结果已有详细报道[10]。
路径二:正庚烷在0价态的单Pt催化剂作用下,脱除氢自由基生成庚基自由基和Pt-H催化活性中心;庚基自由基直接与Pt-H催化活性中心发生化学吸附,进一步发生脱除氢自由基反应,生成庚烯与Pt-H2;庚烯从催化剂表面脱附,而Pt-H2发生脱附反应生成H2和再生的0价态的单Pt催化剂。
对上述2条反应路径的脱氢反应能垒进行计算对比,从而获得一条最优的反应路径。首先将正庚烷分子中的碳原子从分子链一端至另一端依次编号为C(1)、C(2)、C(3)、C(4)、C(5)、C(6)、C(7)。
3.3.1路径一的反应能垒
(1)正庚烷在0价态的单Pt催化剂作用下脱除氢自由基生成庚基自由基
首先计算0价态的单Pt催化剂催化正庚烷分子中的C—H键发生均裂反应生成庚基自由基的反应能垒,计算结果见表1中Step 1部分。

表1 反应路径一中C—H 键均裂的反应能垒(Ea)
由表1 Step 1可见,断开伯碳位的C(1)—H键的反应能垒比仲碳位的C(2)—H键、C(3)—H键、C(4)—H键的反应能垒高约22 kJ/mol,因此反应较难发生。在反应中,仲碳位的C—H键优先发生均裂。
(2)庚基自由基在0价态的单Pt催化剂上脱除氢自由基生成庚烯和氢自由基
由于优先生成仲碳位庚基自由基,选取最易生成的2-庚基自由基为代表,计算自由基碳两边伯碳位上的C(1)—H键与仲碳位上的C(3)—H键均裂时反应能垒的差别,计算结果列于表1 Step 2部分。由此可见,庚基自由基脱除氢自由基生成烯烃时,均裂仲碳位上C(3)—H键的反应能垒比伯碳位上C(1)—H键的反应能垒低27.64 kJ/mol,因此仲碳位的C—H键均裂更易发生。
综上所述,反应路径一中Step 1的最低反应能垒约75.90 kJ/mol,Step 2的最低反应能垒约99.63 kJ/mol。
3.3.2反应路径二
(1)正庚烷在0价态的单Pt催化剂上脱除氢自由基生成庚基自由基
该基元反应的反应能垒列于表2 Step 1部分。由于反应路径二的Step 1与反应路径一中的Step 1为同一反应,因此反应能垒数据及产物分布均相同。生成产物为庚基自由基与具有催化活性的Pt-H催化活性中心。
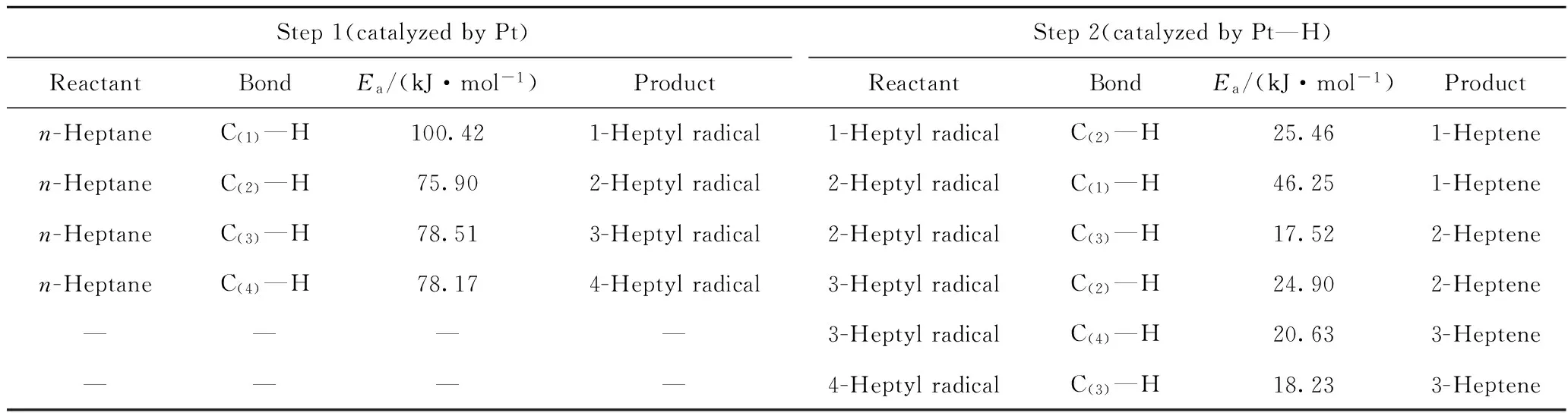
表2 反应路径二中C—H 键均裂的反应能垒(Ea)
(2)庚基自由基直接与Pt-H催化剂活性中心发生化学吸附,进一步发生脱除氢自由基反应,生成庚烯与Pt-H2
选取优先生成的2-庚基自由基为代表进行详细说明,采用过渡态搜索的方法,研究了2-庚基自由基中的C(3)—H键在Pt-H活性中心脱除氢自由基的反应过程,如图3所示。
正庚烷在Pt金属活性中心脱除氢自由基生成2-庚基自由基与Pt-H活性中心后,Pt-H活性中心中的Pt原子拥有1个未成对的自由单电子,2-庚基自由基中断键生成的C(2)自由基原子上同样拥有1个未成对的自由单电子,两者之间存在较强的吸引力。分析认为,2-庚基自由基很难从Pt-H活性中心表面发生脱附,而是直接与Pt-H催化剂活性中心发生化学吸附,进一步发生脱除氢自由基反应。
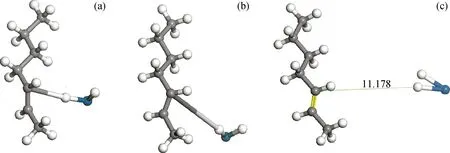
图3 2-庚基自由基在Pt-H活性中心的脱氢反应示意图
图3(a)为经几何优化后的反应物结构,体系结构趋于稳定。对结构进行分析发现,C(3)—H键中的氢原子自动向Pt-H活性中心发生位置偏移,优化后的稳定结构近似于2-庚烯与Pt-H2的化学吸附连接体。由于2-庚烯与Pt-H2两部分均为无单电子的稳定结构,两者间的吸附力比结构稳定前明显减小,从而2-庚烯很容易从Pt-H2表面发生脱附。反应过程中,2-庚基自由基在Pt-H活性中心脱除C(3)—H键中的氢自由基,生成2-庚烯和Pt-H2,反应遵循自由基反应机理,反应能垒为17.52 kJ/mol。
表2 Step 2部分为不同的庚基自由基在Pt-H活性中心催化下脱除氢自由基的反应能垒。仲碳位C—H键的均裂反应能垒在17.52~25.46 kJ/mol范围,而伯碳位C(1)—H键的均裂反应能垒为46.25 kJ/mol,相比而言仲碳位C—H键更容易发生反应。
从能量角度分析,在反应路径二中,正庚烷分子不同仲位碳原子(C(2)、C(3)、C(4))的热力学性质近似相同。在Step 1,生成2-庚基自由基、3-庚基自由基、4-庚基自由基,其中优先生成2-庚基自由基。在Step 2,1-庚基自由基生成产物为1-庚烯;2-庚基自由基生成产物包括2-庚烯和3-庚烯,其中优先生成2-庚烯;3-庚基自由基的与自由基碳相邻的两侧仲碳原子(C(2)、C(4))性质近似,脱氢生成2-庚烯与3-庚烯的概率近似; 4-庚基自由基由于自身为对称结构,因此脱氢产物为3-庚烯。
综上所述,反应路径二中,Step 1的最低反应能垒约75.90 kJ/mol,Step 2的最低反应能垒为17.52 kJ/mol。正庚烷脱氢生成正庚烯的最终产物主要为2-庚烯和3-庚烯,其中优先生成2-庚烯。
(3)由于反应路径一与反应路径二中庚烯从催化剂表面的脱附反应及H2在Pt催化剂表面的脱附反应原理相同,在此不进行详细叙述。
3.3.3反应路径一与反应路径二的对比
从能量角度而言,基元反应优先沿着反应能垒较低的路径进行。而通过对比2条反应路径中各基元反应的反应能垒,可以获得最优反应路径。
对于Step 1,由于正庚烷都是在0价态的Pt原子催化下进行脱除氢自由基反应,2条反应路径相同,反应能垒相同。
对于Step 2,在反应路径一中,庚基自由基在0价态的Pt原子催化下进行脱除氢自由基反应,仲碳位C—H键的脱氢反应能垒约为99.63 kJ/mol;在反应路径二中,庚基自由基在Pt-H活性中心催化下进行脱除氢自由基反应,仲碳位C—H键的反应能垒约为17.52 ~ 25.46 kJ/mol。反应路径二比反应路径一中基元反应的反应能垒大幅度降低约75 kJ/mol,反应极易发生,说明庚基自由基优先在Pt-H活性中心继续发生反应并生成庚烯。
对比2条反应路径中的Step 1与Step 2得出,反应路径二为最优反应路径。该反应路径中最大反应能垒为75.89 kJ/mol,其中Step 1为速控步骤。同时优先生成2-庚烯。
由此得出正庚烷脱氢生成烯烃反应的最优反应路线为,(1) 正庚烷首先在0价态的Pt原子表面发生化学吸附;(2) 正庚烷在0价态的单Pt催化剂催化下发生脱除氢自由基反应,生成庚基自由基和Pt-H 活性中心;(3) 庚基自由基与同时生成的Pt-H活性中心发生化学吸附;(4) 庚基自由基在Pt-H活性中心发生脱氢自由基反应,生成产物庚烯和Pt-H2;(5) 庚烯从Pt-H2表面发生脱附,而Pt-H2发生脱附反应生成H2和再生的0价态的单Pt催化剂。
3.4Pt催化剂催化正庚烷脱氢生成烯烃路径的实验验证
采用Pt-γ-Al2O3-Cl催化剂,依次在370、400、450、500℃下进行正庚烷催化重整反应。测定反应产物的液相组成,并采用单体烃PONA组成分析方法分析液相产物中庚烯所占的质量分数,结果列于表3。
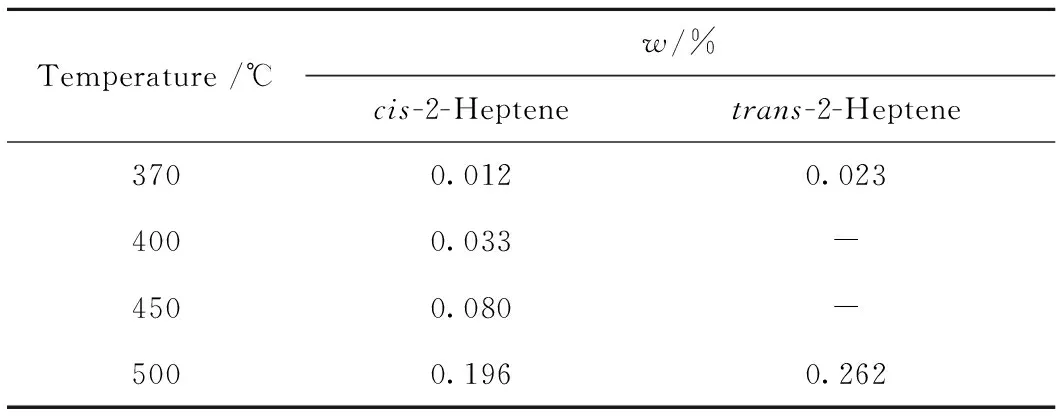
表3 Pt催化剂催化正庚烷脱氢反应液相产物中庚烯组成
从表3看出,在常规催化重整实验过程中,正庚烷脱氢生成庚烯的产物组成中主要由顺-2-庚烯和反-2-庚烯组成,而未发现3-庚烯和1-庚烯的存在。分析认为,反应过程中可能生成极少量3-庚烯和1-庚烯,但色谱检测器很难进行识别。最终,从实验角度证明,正庚烷脱氢生成正庚烯的反应过程中优先生成2-庚烯。
4结论
(1)Pt原子在脱氢反应中生成的Pt-H活性中心具有吸取单电子的能力,具有较强的脱除氢自由基的催化作用。
(2)Pt催化剂催化正庚烷脱氢生成烯烃存在2条反应路径,反应路径二为最优反应路径。正庚烷首先在0价态的Pt原子表面发生化学吸附,随后发生脱除氢自由基反应生成庚基自由基和Pt-H活性中心,最低反应能垒为75.89 kJ/mol;庚基自由基直接与Pt-H催化剂活性中心发生化学吸附,进一步发生脱除氢自由基反应,生成庚烯与Pt-H2,最低反应能垒为17.52 kJ/mol;庚烯从Pt-H2表面发生脱附,而Pt-H2发生脱附反应生成H2和再生的 0价态的单Pt催化剂。
(3)反应路径二中,正庚烷在0价态的单Pt催化剂催化下发生脱除氢自由基反应生成庚基自由基和Pt-H活性中心的反应为速控步骤,同时优先生成2-庚烯。实验证明,正庚烷脱氢生成正庚烯的反应过程中优先生成2-庚烯。
参考文献
[1] BURTRON H D. Alkane dehydrocyclization mechanism[J].Catalysis Today, 1999, 53(3):443-516.
[2] 杨锡尧, 潘韫, 庞礼, 等. 常压下Pt/Al2O3催化剂的正庚烷脱氢芳构化活性中心[J].催化学报, 1985, 6(3):288-291.(YANG Xiyao, PAN Yun, PANG Li, et al. The active site of Pt/Al2O3catalyst for the dehydrogenation ofn-heptane at normal pressure[J].Journal of Catalysis, 1985, 6(3):288-291.)
[3] 杨锡尧, 刘燕萍, 陆新, 等. Pt-Al2O3的烃类催化转化活性中心性质[J].燃料化学学报, 1985, 13(2):97-105.(YANG Xiyao, LIU Yanping, LU Xin, et al. The nature of the active sites in catalysts of Pt-Al2O3for hydrocarbon conversion[J].Journal of Fuel Chemistry and Technology, 1985, 13(2):97-105.)
[4] 方大伟, 马爱增, 潘锦程. Pt/ ZrO2-γ-Al2O3催化剂芳构化反应性能研究[J].石油炼制与化工, 2008, 39(3):28-33.(FANG Dawei, MA Aizeng, PAN Jincheng. Study on aromatization performance of Pt/ ZrO2-γ-Al2O3catalyst[J].Petroleum Processing and Petrochemicals, 2008, 39(3):28-33.)
[5] MILLS G A, HEINEMANN H, MILLIKEN T H, et al. Catalytic mechanism[J].Industrial Engineering & Chemistry, 1953, 45(1):134-224.
[6] KEULEMANS A I M, VOGE H. Reactivities of naphthenes over a platinum reforming catalyst by a gas chromatographic technique[J].Journal of Physical Chemistry, 1959, 63(4):476-480.
[7] 刘伟成, 田志坚, 徐竹生. 正癸烷脱氢生成直链单烯烃的热力学分析[J].石油学报(石油加工), 2001, 17(4):39-43.(LIU Weicheng, TIAN Zhijian, XU Zhusheng. A thermodynamic analysis on dehydrogenation ofn-decane to decene[J].Acta Petrolei Sinica (Petroleum Processing Section), 2001, 17(4):39-43.)
[8] 张高勇, 刘骥, 唐鸿鑫. 长链烷烃脱氢主反应及其失活过程表观动力学研究[J].燃料化学学报, 1983, 11(2):48-59.(ZHANG Gaoyong, LIU Ji, TANG Hongxin. Kinetics of catalytic dehydrogenation of long chain paraffins and its deactivation[J].Journal of Fuel Chemistry and Technology, 1983, 11(2):48-59.)
[9] 康保安, 唐鸿鑫, 张高勇, 等. 长链烷烃脱氢动力学的研究[J].日用化学工业, 1986, (3):1-6.(TANG Baoan, TANG Hongxin, ZHANG Gaoyong, et al. Research into the dynamics of long-chain alkenes dehydrogenation[J].Daily Chemical Industry, 1986, (3):1-6.)
[10] 于宁, 龙军, 周涵, 等. 正庚烷脱氢生成烯烃反应的分子模拟[J].石油学报(石油加工). 2013, 29(2):181-185.(YU Ning, LONG Jun, ZHOU Han, et al. Study on molecule simulation of dehydrogenation ofn-Heptane to produce olefins[J].Acta Petrolei Sinica (Petroleum Processing Section), 2013, 29(2):181-185.)
[11] YAO H C, SIEG M, PLUMMER Jr H K. Surface interaction in the Pt/γ- Al2O3system[J].Journal of Catalysis, 1979, 59(3): 365-374.
[12] 彭红建, 谢佑卿, 聂跃庄. 电催化剂Pt的电子结构和催化性能[J].中南大学学报(自然科学版), 2007, 38(1):98-101.(PENG Hongjian, XIE Youqing, NIE Yuezhuang. Electronic structures and catalytic performance of Pt-electrocatalyst[J].Journal of Central South University (Science and Technology), 2007, 38(1):98-101.)
[13] JERZY S, BARTOMIEJ S. Adsorption of C7 hydrocarbons in microporus materials: Molecular modeling[J].Microporous and Mesoporous Materials, 2004, 76(1):247-254.
[14] MAIER G, COROLLEUR C, JUTTARD D, et al. Comments on a dispersion effect in hydrogenolysis of methylcyclopentane and isomerization of hexanes over supported platinum catalysts[J].Journal of Catalysis, 1971, 21(2):250-253.
[15] TAYLOR W F, SINFELT J H, YATES D J C. Catalysis over supported metal Ⅳ Ethane hydrogenolysis over dilute nickel catalysts[J].Journal of Physical Chemistry, 1965, 69(11):3857-3863.
[16] CARTER J L, CUSUMANO J A, SINFELT J H. Catalysis over supported metal Ⅴ The effect of crystallite size on the catalytic activity of nickel catalysts[J].Journal of Physical Chemistry, 1966, 70(7):2257-2263.
Simulation of the Reaction Mechanism of Dehydrogenation of n-Heptane to Produce Olefins
YU Ning, LONG Jun, ZHOU Han, MA Aizeng, DAI Zhenyu
(ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China)
Abstract:The dehydrogenation of n-heptane to olefins in the process of catalytic reforming was studied by density functional theory (DFT) quantum chemical methods. Two different reaction approaches were compared. It is concluded that the Pt-H reactivity center yielded from the Pt atom during the dehydrogenation is capable of capturing single electron, which has the strong catalytic capability to capture H radical. During the reaction, n-heptane was first absorbed on the surface of the zero-valence Pt, and then the H radical is removed, yielding the heptyl radical and the Pt-H reactivity center. 2-heptyl radical was produced with priority due to the minimum barrier energy of 75.89 kJ/mol. Heptyl radical is chemically absorbed to the Pt-H reactivity center directly, which loses H radical subsequently, resulting in the formation of heptane and Pt-H2. 2-Heptene is produced with priority due to the minimum barrier energy of 17.52 kJ/mol. Heptane is desorbed on the surface of Pt-H2, and H2and the regenerated zero-valence Pt are obtained by desorption of Pt-H2. The maximum barrier energy of this reaction approach is 75.89 kJ/mol. It is proved that heptane gives the priority to producing 2-heptene during the dehydrogenation.
Key words:n-heptane; dehydrogenation of alkanes; Pt; reaction mechanism; molecule simulation
收稿日期:2015-03-26
基金项目:中国石油化工股份有限公司项目(R14056)资助
文章编号:1001-8719(2016)03-0437-07
中图分类号:TE624
文献标识码:A
doi:10.3969/j.issn.1001-8719.2016.03.001
通讯联系人: 于宁,男,博士,从事催化重整反应机理的研究;Tel:010-82368768;E-mail:yuning.ripp@sinopec.com
