肝豆片质量标准改进研究
2016-06-27臧恒昌
卜 伟,臧恒昌
(1.山东大学药学院,山东 济南 250012;2.合肥立方制药股份有限公司,安徽 合肥 230088)
肝豆片质量标准改进研究
卜伟1,2,臧恒昌1
(1.山东大学药学院,山东 济南250012;2.合肥立方制药股份有限公司,安徽 合肥230088)
摘要:昌目的增加对肝豆片处方中药材质量控制方法,全面提高肝豆片质量标准,更有效地控制产品的质量。方法建立高效液相色谱法对处方中君药大黄进行含量测定,并对处方中臣药黄连、金钱草建立薄层色谱鉴别方法。结果黄连鉴别以甲苯-异丙醇-乙酸乙酯-甲醇-水(6∶1.5∶3∶1.5∶0.5)为展开剂,以浓氨水作为饱和溶液,置紫外灯365 nm下检视;金钱草鉴别以甲苯-甲酸乙酯-甲酸(10∶8∶1)为展开剂,以3%三氯化铝乙醇溶液为显色剂,置紫外灯365 nm下检视;大黄含量测定大黄素在8.48~25.44 mg·L-1的浓度范围,峰面积A与浓度C呈现良好的线性关系,相关系数为0.999 4,回收率为95.8%,RSD=0.8%(n=6)。结论方法准确、快速、重现性好,质量标准得到改进。
关键词:大黄素;色谱法,高压液相;色谱法,薄层;黄连碱;金钱草;质量控制
肝豆片主要由大黄、黄连、金钱草等六味中药组成。具有清热解毒,通腑利湿功效。临床上主要用于治疗肝豆状核变性。肝豆片对改善患者构音障碍、肌强直、震颤等症状具有一定作用,避免了金属络合剂驱铜治疗易产生的过敏反应、骨髓抑制、肾脏损害及类红斑狼疮等副作用[1]。
目前,肝豆片质量标准(安徽省食品药品监督管理局医疗机构制剂标准,标准号为皖Q/WS-ZJ(Z-005)-2012)仅对处方中的大黄、三七进行鉴别,缺少定量分析方法,且处方中其他药材缺乏质量控制手段,因此,有必要对该品种质量标准进行改进,以更好地控制产品质量。本文通过查阅相关文献和实验研究,对该处方中君药大黄建立含量测定方法,结果表明此方法准确、稳定、可靠;同时对处方中臣药黄连、金钱草建立薄层色谱鉴别方法,结果表明该方法可行、有效、专属性好。通过这些质量标准的改进,有效地提升了肝豆片质量控制水平。
1仪器和试剂
1.1仪器Shimadzu LC-20AD高效液相色谱仪(岛津)、LCsolutions(岛津)、电子分析天平(梅特勒-托利多)、超声波清洗器(天津奥特赛恩斯有限公司)、硅胶G薄层板。
1.2试剂含量测定项中甲醇为色谱纯;盐酸小檗碱、槲皮素、山奈素、大黄素为对照品(中国药品生物制品检定研究院);供试品为肝豆片(合肥立方制药股份有限公司自制);其余试剂均为分析纯;水为二次蒸馏(自制)。
2肝豆片中黄连的鉴别
2.1方法的建立查阅相关资料[2-4],并参照《中国药典》2010版一部“黄连上清片”及“黄连”项下黄连鉴别方法[5]初步选定本品中黄连的鉴别方法,并对其中的展开剂进行优选,结果见表1。
结合以上展开剂优选的结果,初步确定本品中黄连的鉴别方法如下:(1)供试品溶液制备:取本品粉末1.2 g,加甲醇25 mL,超声处理30 min,滤过,取滤液作为供试品溶液。(2)对照品溶液制备:称取盐酸小檗碱对照品2.0 mg,加甲醇4 mL溶解作为对照品溶液。
检测方法:照薄层色谱(《中国药典》2010年版一部附录Ⅵ B)法测定,吸取供试品溶液、对照品溶液各1 μL,分别点于同一硅胶G薄层板上,以甲苯-异丙醇-乙酸乙酯-甲醇-水(6∶1.5∶3∶1.5∶0.5)为展开剂,另一槽加入等体积的浓氨水,预饱和10 min后,展开,取出,晾干,置紫外灯365 nm下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。
2.2方法的验证
2.2.1专属性(1)阴性对照溶液:取阴性对照样品(按处方比例和处方工艺制备不加黄连)1.2 g,加甲醇25 mL,超声处理30 min,滤过,取滤液作为阴性对照溶液。(2)供试品溶液:取本品1.2 g,加甲醇25 mL,超声处理30 min,滤过,取滤液作为供试品溶液。(3)对照品溶液制备:称取盐酸小檗碱对照品2.3 mg,加甲醇4 mL溶解作为对照品溶液。
分别吸取上述溶液各1 μL,点于同一硅胶G薄层板上,按“2.1”项中建立的方法进行检测。结果表明,阴性对照色谱中,在与对照品色谱相应的位置上,无任何斑点。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,其他位置斑点与主斑点分离完全。表明制剂中其他组分对黄连的鉴别无干扰,所建立的鉴别检验方法专属性较好。薄层图谱见图1。
2.2.2耐用性分别选用青岛海洋化工厂分厂、烟台江友硅胶开发有限公司、青岛胜海精细硅胶化工有限公司三家薄层板的厂家进行鉴别实验,所获得的薄层图谱分离度均较好、主斑点斑型集中。同时对点样方式分别采用圆点状和窄细条带状进行实验,实验结果证明点样方式对薄层图谱的分离度影响较小。
3肝豆片中金钱草的鉴别
3.1方法的建立查阅相关文献[6-8],并参照《中国药典》2010版一部“金钱草”项下鉴别方法进行实验,取本品进行薄层色谱鉴别,薄层色谱中斑点清晰、分离度较好、斑型集中,故按照此方法建立本品中金钱草的鉴别方法,方法如下。
3.1.1供试品溶液的制备取本品粉末4.5 g,加80%甲醇50 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加水10 mL使溶解,用乙醚振摇提取2次,每次10 mL,弃去乙醚液,水液加稀盐酸10 mL,置水浴中加热回流1 h,取出,冰浴10 min后,用乙酸乙酯振摇提取2次,每次20 mL,合并乙酸乙酯液,用水30 mL洗涤,弃去水液,乙酸乙酯液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。
3.1.2对照品溶液的制备另取槲皮素对照品、山奈素对照品,加甲醇制成每1 mL各含0.5 mg的混合溶液,作对照品溶液。
3.1.3检测方法照薄层色谱法(《中国药典》2010年版一部附录Ⅵ B)测定,吸取供试品溶液3 μL,对照品溶液2 μL,分别点于同一硅胶G薄层板上,以甲苯-甲酸乙酯-甲酸(10∶8∶1)为展开剂,展开,取出,晾干,喷以3%三氯化铝乙醇溶液,在105℃加热数min,置紫外灯(365 nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。

表1 黄连鉴别方法展开剂优选结果
3.2方法的验证
3.2.1专属性(1)阴性对照溶液制备:取阴性对照样品(按处方比例和处方工艺制备不加金钱草)4.5 g,按照上述供试品溶液制备方法制备阴性对照溶液。(2)供试品溶液制备:取本品4.5 g,按照变上述供试品溶液制备方法制备供试品溶液。(3)对照品溶液制备:称取槲皮素对照品2.51 mg、山奈素对照品2.55 mg,置5 mL容量瓶中。加甲醇溶解并稀释至刻度,摇匀,作为对照品溶液。
分别吸取上述三种溶液,点于同一硅胶G薄层板上,按“3.1”项中建立的方法进行检测。结果表明,对照品色谱中,两个主斑点清晰可辨,分离完全。阴性对照色谱中,在与对照品色谱相应的位置上,无任何斑点。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,其他位置斑点与主斑点分离完全。制剂中其他组分对金钱草的鉴别无干扰,所建立的鉴别检验方法专属性较好。薄层图谱见图2。


注:1.缺味阴性样品;注:1.缺味阴性样品;
2.盐酸小檗碱对照品; 2.槲皮素和山奈素混合对照品;
3.肝豆片供试品。3.肝豆片供试品。
图1黄连TLC图图2金钱草TLC图
3.2.2耐用性分别选用青岛海洋化工厂分厂、烟台江友硅胶开发有限公司、青岛胜海精细硅胶化工有限公司三家薄层板的厂家进行鉴别实验,所获得的薄层图谱分离度均较好、主斑点斑型集中。同时对点样方式分别采用圆点状和窄细条带状进行实验,实验结果证明点样方式对薄层图谱的分离度影响较小。
4含量测定
4.1标准限度的确定统计分析历年来采购的大黄素在大黄药材与肝豆片制剂中含量的对应关系,结果见表2,并结合大黄药材中大黄素的内控指标,药典中其他类似品种的含量限度及自身测定的结果,制定本品的含量限度。
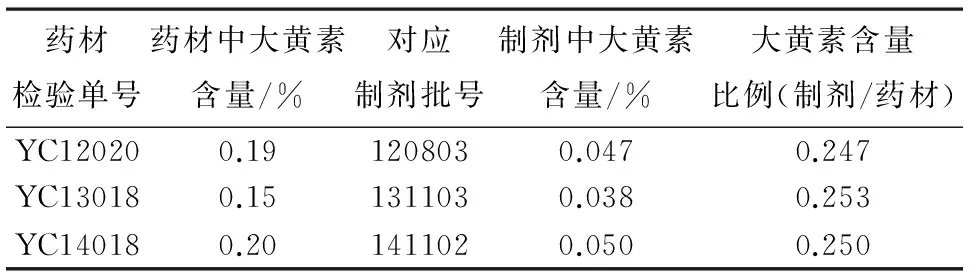
表2 大黄药材与肝豆片药材含量对应关系
大黄素含量比例(制剂/药材)平均值为0.25,大黄药材中大黄素内控指标为0.11%,折算制剂中大黄素限度为0.0275%,折合每片含大黄素0.124 mg,并参考药典中其他类似品种限度,将本品限度定为:本品每片含大黄以大黄素(C15H10O5)计,不得少于0.13 mg。
4.2方法的建立查阅相关文献[9-11],并进行相关的对比实验,最终建立本品的含量测定方法如下:
照高效液相色谱法(中国药典2010版一部附录ⅥD)测定。
色谱条件与系统性适用性试验:以十八烷基键合硅胶为填充剂;以甲醇-0.1%磷酸溶液(85∶15)为流动相;流速:1.0 mL·min-1;检测波长:254 mm;柱温:30℃;理论板数按大黄素峰计算应不低于3 000。
对照品溶液的制备:精密称取大黄素对照品16 mg置100 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,取上述溶液1 mL置10 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得。
供试品溶液的制备:取本品适量研细,取约2.7 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,称定重量,加热回流1 h,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过。精密量取续滤液10 mL,置具塞锥形瓶中,水浴挥干溶剂,加8%盐酸溶液10 mL,超声2 min,再加三氯甲烷10 mL,加热回流1 h,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次(10、10、15 mL),合并三氯甲烷液置60℃水浴锅上挥干,残渣加甲醇使溶解,转移置10 mL量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液作为供试品溶液。
测定法:分别精密吸取上述对照品溶液和供试品溶液各10 μL注入液相色谱仪,测定,按外标法以峰面积计算,即得。本品每片含大黄以大黄素(C15H10O5)计,不得少于0.13 mg。
4.3方法的验证
4.3.1专属性(1) 空白溶剂:取溶剂甲醇,采用0.45 μm的微孔滤膜过滤,即得。(2) 阴性对照溶液:取阴性对照样品(按处方比例和处方工艺制备不加大黄)2.7 g,按照上述供试品溶液的制备方法,制备阴性对照溶液。(3) 对照品溶液:精密称取大黄素对照品16 mg置100 mL容量瓶中,加甲醇溶解并定量稀释至刻度,取上述溶液1 mL至10 mL容量瓶中,加甲醇溶解并定量稀释至刻度,摇匀,即得。(4) 供试品溶液:取本品2.7 g,按照上述供试品溶液的制备方法,制备供试品溶液。
分别精密吸取上述溶液各10 μL,注入液相色谱仪,按照“4.2”项下色谱条件进行测定,检验结果见图3。
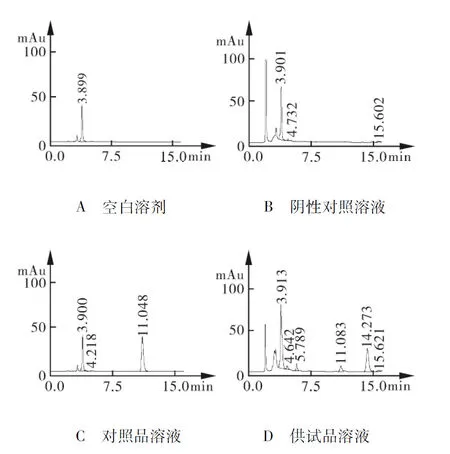
图3肝豆片大黄素含量测定HPLC色谱图
对照品溶液中大黄素色谱峰保留时间为11 min,空白溶剂色谱图中在保留时间3.9 min有色谱峰,阴性对照色谱图中在2.0、3.8、3.9 min有色谱峰,在与对照品主峰位置11 min无色谱峰;供试品溶液色谱图中在相应位置出现色谱峰,大黄素峰的理论板数为11 189,与相邻位置色谱峰的分离度分别为8.63、6.63。
结果表明:溶剂及制剂中其他组分对大黄的含量测定无干扰,所建立的含量测定方法专属性良好。
4.3.2线性与范围精密称取大黄素对照品16.96 mg置100 mL容量瓶中,加甲醇溶解并定量稀释至刻度,分别精密量取上述溶液0.5、0.75、1.0、1.25、1.5 mL分别至10 mL容量瓶中,加甲醇溶解稀释至刻度,摇匀,即得。分别精密量取上述5种溶液10 μL,注入液相色谱仪,记录色谱图,以浓度(mg·L-1)对峰面积进行线性回归,结果大黄素在8.48~25.44 mg·L-1的浓度范围,峰面积A与浓度C呈现良好的线性关系,相关系数为0.999 4。
4.3.3准确度对照品溶液制备:精密称取大黄素对照品16.96 mg置100 mL容量瓶中,加甲醇溶解并定量稀释至刻度,取上述溶液1 mL至10 mL容量瓶中,加甲醇溶解并定量稀释至刻度,摇匀,即得。
对照品储备液的制备:精密称取大黄素对照品(含量:98.7%)10.05 mg置100 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得。
精密称取已知大黄素含量(批号:141102,含量:0.50 mg·g-1)的供试品粉末0.8 g,分别置9个具塞锥形瓶中,其中三个锥形瓶中分别精密加入上述对照品储备液2 mL,另外三个锥形瓶中分别精密加入上述对照品储备液4 mL,最后三个锥形瓶中分别精密加入上述对照品储备液6 mL,再按照拟定的含量测定方法项下供试品溶液的制备方法,制备即得。
精密吸取上述溶液各10 μL,注入液相色谱,按拟定的色谱条件进行测定,记录色谱图,计算回收率。结果见表3。结果表明:大黄素的平均回收率为97.2%>95%,说明本方法的准确度良好。
4.3.4重复性精密称取本品粉末2.7 g,共6份,按“4.2”项下供试品溶液的制备方法,制备成6份供试品溶液。精密称取大黄素对照品16 mg,置100 mL容量瓶中,加甲醇溶解并定量稀释至刻度,取上述溶液1 mL至10 mL容量瓶中,加甲醇溶解并定量稀释至刻度,摇匀,即得对照品溶液。分别精密量取对照品溶液和供试溶液各10 μL,注入液相色谱仪,记录色谱图,按外标法以峰面积计算供试溶液的浓度,计算大黄素的测定浓度RSD(%)为1.9%,小于2.0%,说明本方法测定大黄素重复性较好。

表3 大黄素回收率检查结果
4.3.5中间精密度照“重复性”项下操作,由不同人配制相同浓度的供试溶液,并由该操作者进样分析。按照变更后质量标准中含量测定方法,测定供试溶液中大黄素的含量,计算RSD为0.8%,小于5.0%,说明此方法的中间精密度良好。
4.3.6稳定性按照变更后质量标准中含量测定项下方法配制对照品溶液和供试品溶液。按拟定的色谱条件在0、2、4、6、8、10、12、14、16和24 h进样,记录其峰面积,对照品溶液和供试品溶液中大黄素峰面积的RSD分别为0.14%和0.64%,表明对照品溶液和供试品溶液在24 h内稳定性良好。
4.3.7耐用性条件参数变化对分析方法的影响将通过改变检测波长、流速、柱温、流动相比例、更换不同厂家相同规格色谱柱评估。考察变动参数:检测波长分别为252、256 nm;流速分别为0.8、1.2 mL·min-1;柱温分别设置为25℃、35℃;不同色谱柱型号:Welth XB C18250 mm×4.6 mm,5 μm;流动相:甲醇-0.1%磷酸溶液(80∶20),甲醇-0.1%磷酸溶液(90∶10)。实验结果证明,改变流速、更改色谱柱、改变流动相比例、改变柱温、改变波长等条件,对含量测定结果影响较小,说明该方法的耐用性较好。
5讨论
本方中君药为大黄,主要利用其泻下作用。以大黄素为本品的含量控制指标,可以反映该制剂的质量。本文中选取高效液相色谱法进行大黄素的含量测定替代原有质量标准中大黄的薄层鉴别,能更有效地控制产品的质量。
本文参照了相关文献并结合药典中类似品种的鉴别方法,并进行相应的改进,建立了本品的黄连和金钱草的鉴别方法。斑型较好,分离完全,斑点清晰,专属性较好,能够排除其他成分的干扰。
参考文献
[1]胡文彬,杨任民.肝豆片I号对肝豆状核变性患者微量元素排泄的影响[J].中国临床康复,2003,7(30):4165.
[2]薛非凡,曾丽,王丽玉,等.消渴灵片质量标准研究 [J].中国药师,2013,16(11):1641-1644.
[3]孙建彬,王欣,覃瑶,等.黄连及其炮制品和黄连须的薄层鉴别 [J].华西药学杂志,2014,29(6):675-677.
[4]肖丽和,关潇滢,韩东岐,等.万氏牛黄清心片质量标准研究[J].中国药师,2013,16(2):239-241.
[5]国家药典委员会.中国药典(一部)[S].北京:中国医药科技出版社,2010:130-131.
[6]马军花,邱宏聪,陈明生,等.复方金钱草颗粒的质量控制研究[J].现代药物与临床,2014,29(4):381-384.
[7]朱雪莲,方琦.复方金钱草膏的制备及薄层鉴别[J].世界中医药,2013,8(3):332-333.
[8]李兵.利胆退黄合剂的薄层鉴别[J].临床合理用药杂志,2012,5(8):92.
[9]黄尹,李国峰.不同产地虎杖中各成分含量的比较 [J].中医药信息,2015,32(5):68-71.
[10] 徐煜纯,谢明容,刘少峰,等.HPLC法测定壮药铁包金中大黄素、大黄酚的含量[J].广东药学院学报,2014,30(2):169-172.
[11] 张强,冯华,吴绍维,等.HPLC色谱法测定首乌延寿片中大黄素的含量[J].医药前沿,2015,5(12):82-83.
Study on the improvement of the quality standard of Gandou Tablets
BU Wei1,2,ZANG Heng-chang1
(1.SchoolofPharmaceuticalSciencesShandongUniversity,Ji′nan,Shandong250012,China;2.HefeiLifeonPharmaceuticalCo.,Ltd.,Hefei230088,China)
Abstract:ObjectiveIncrease in hepatic bean prescription medicines quality control methods,quality standards,improving liver beans more effectively control the quality of the products.MethodEstablish a high performance liquid chromatography determination of liver beans tablets in the content of rhubarb;Using thin-layer chromatography on identification of rhizoma coptidis,lysimachia christinae;Using high-performance liquid chromatographic method for the determination of emodin.ResultsThe identification of the identification of the root of the (6∶1.5∶3∶1.5∶0.5) was used as the solvent,and the concentration of the solution was in the light of the concentration of 365 nm;Hristina loosestrife herb with toluene ethyl formate and formic acid 10∶8∶1 as expansion agent,with 3% solution of aluminium chloride in ethanol as chromogenic agent,placed under ultraviolet light at 365 nm view;Emodin 8.48~25.44 mg·L-1concentration range,the peak concentration of area a and c presents a good linear relationships,the correlation coefficient is 0.999 4,the returns-ratio is 95.8%,RSD=0.8%(n=6).ConclusionThe method is accurate,rapid and with good reproducibility,the quality standards are improved.
Key words:Emodin;Chromatography,High Pressure Liquid;Chromatography,Thin Layer;Coptisine;Lysimachia Christinae;Quality Control
作者简介:卜伟,男,硕士研究生 通信作者:臧恒昌,男,教授,博士生导师,研究方向:多糖类药物研究、药品生产工艺优化及在线过程分析与过程控制, E-mail:zanghcw@126.com
doi:10.3969/j.issn.1009-6469.2016.05.007
(收稿日期:2016-02-12,修回日期:2016-04-07)
