HPLC法测定劳拉西泮片有关物质的含量
2016-06-27孙爱华胡文辉
孙爱华,胡文辉
(1.黄冈市中心医院药剂科,湖北 黄冈 438000;2.鄂州市中心血站,湖北 鄂州 436000)
HPLC法测定劳拉西泮片有关物质的含量
孙爱华1,胡文辉2
(1.黄冈市中心医院药剂科,湖北 黄冈438000;2.鄂州市中心血站,湖北 鄂州436000)
摘要:目的建立劳拉西泮片的含量及有关物质的HPLC方法。方法以十八烷基硅烷键合硅胶作为填充剂的ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm)为色谱柱,流动相:0.05 mol·L-1的磷酸二氢铵(含0.5%的三乙胺,用磷酸调pH至2.5)∶甲醇∶乙腈=35∶35∶30;流速:1.0 mL·min-1;检测波长:235 nm;进样量:10 μL;柱温:30℃。结果劳拉西泮峰及各杂质峰均能良好分离。杂质A浓度与峰面积在1.5 μg·L-1~8.048 mg·L-1内线性关系良好。杂质B浓度与峰面积在15.0 μg·L-1~8.224 mg·L-1内线性关系良好。杂质C浓度与峰面积在1.7 μg·L-1~8.144 mg·L-1内线性关系良好;杂质D浓度与峰面积在2.5 μg·L-1~8.032 mg·L-1内线性关系良好;杂质E浓度与峰面积在3.0 μg·L-1~8.032 mg·L-1内线性关系良好。测定样品含量平均值为97.9%,有关物质均符合要求。结论该方法快速、简单,稳定,重现性好,可作为劳拉西泮片的含量及有关物质的检测方法。
关键词:劳拉西泮;色谱法,高压液相
《美国药典》35版[4]均收录了劳拉西泮原料和其片剂、口服溶液剂及注射剂。两版药典中的劳拉西泮原料药部分,列出了5个有关物质,有两个工艺杂质,分别是杂质A和杂质B,其中杂质B即是工艺杂质又是降解产物,所以对杂质B进行了严格控制,对杂质A则未做具体要求。《欧洲药典》[5]只收载了劳拉西泮原料药,并用薄层色谱法对有关物质杂质A和杂质B进行测定,但未做具体要求。《中国药典》2015版[6]均收载了劳拉西泮和劳拉西泮片,原料药考察了杂质Ⅰ(即杂质B)和杂质Ⅱ(即杂质C),片剂只考察了杂质Ⅱ(即杂质C)。
为了提高劳拉西泮原料药及制剂的质量,根据其生产工艺条件及降解特点,并结合强制破坏试验的结果,最终确定了劳拉西泮原料药及制剂的有关物质,包括杂质A、杂质B、杂质C、杂质D和杂质E(结构见图1)。本文主要建立了一种可以同时测定劳拉西泮片剂和其有关物质检测的HPLC方法,经验证该方法操作简便、灵敏和专属性强。
1仪器与试药
Agilent 1260型高效液相色谱仪,紫外检测器(Agilent)及安捷伦色谱工作站,电子分析天平(AE230S 梅特勒-托利多上海有限公司),SB5200超声仪(必能信超声上海公司)。色谱纯乙腈,色谱纯甲醇,超纯水,磷酸二氢铵(分析纯,天津市科密欧化学试剂有限公司),三乙胺(分析纯,国药集团化学试剂有限公司),磷酸(分析纯,国药集团化学试剂有限公司)。劳拉西泮对照品(中国药品生物制品检定所,批号171253-200401),杂质A、B、C、D、E为自制。劳拉西泮片(泰国大西洋制药有限公司,批号:140524),规格1.0 mg。
2方法和结果
2.1色谱条件色谱柱:ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm);流动相:0.05 mol·L-1的磷酸二氢铵(含0.5%的三乙胺,用磷酸调pH至2.5)∶甲醇∶乙腈=35∶35∶30;流速:1.0 mL·min-1;检测波长:235 nm;进样量:10 μL;柱温:30℃。
2.2溶液配制取劳拉西泮片对照品20片,研细,取细粉适量(约相当于劳拉西泮20 mg),置于100 mL容量瓶中,加入适量的流动相溶解,超声10 min,放冷,补加流动相至刻度,摇匀,过滤,滤液作为供试品溶液。精密量取1.0 mL,置50 mL量瓶中,用流动相稀释至刻度,摇匀,作为对照品溶液。
精密称取劳拉西泮对照品5.06 mg、杂质A对照品5.03 mg、杂质B对照品5.14 mg、杂质C对照品5.09 mg、杂质D对照品4.97 mg、杂质E对照品5.02 mg,分别置于25 mL容量瓶中,加入乙腈溶解并稀释至刻度,摇匀,作为劳拉西泮和各杂质对照品的贮备溶液。
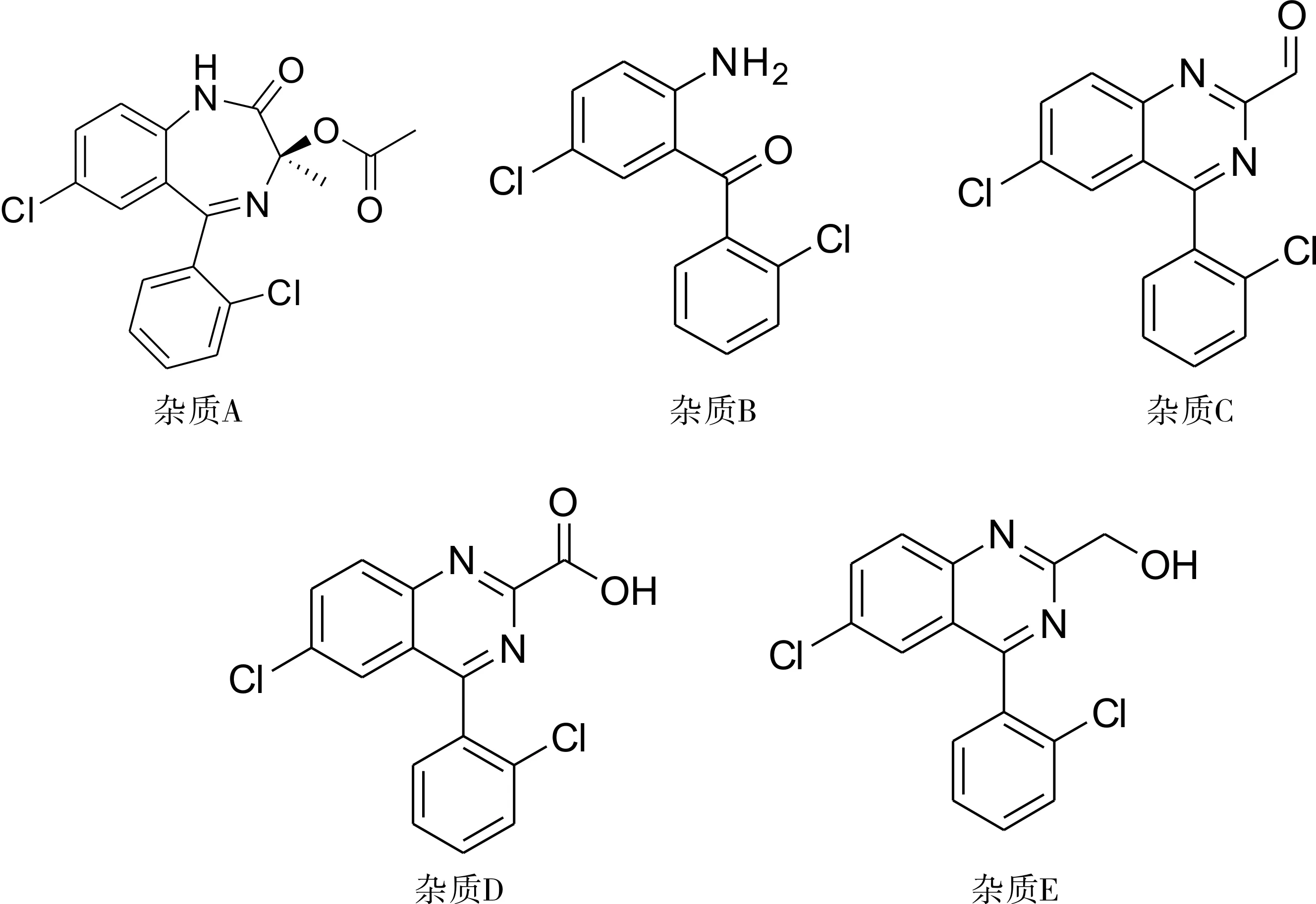
注:杂质A.3(RS)-7-氯-5-(2-氯苯基)-1,3-二氢-乙酰氧基-2H-苯二氮-2-酮;杂质B.(2-氨基-5-氯苯基)(2-氯苯基)甲酮;杂质C.6-氯-4-(2-氯苯基)-喹唑啉-2-醛;杂质D.6-氯-4-(2-氯苯基)-喹唑啉甲酸;杂质E.6-氯-4-(2-氯苯基)-喹唑啉甲醇。
图1劳拉西泮原料药及制剂的有关物质结构图
2.3方法学考察
2.3.1系统适用性试验分别精密量取上述劳拉西泮及各杂质对照品的贮备溶液1.0 mL,置于10 mL的容量瓶中,加入流动相溶解并稀释至刻度,作为系统适用性溶液。精密量取10 μL注入高效液相色谱仪,记录色谱图,各峰理论板数要求不低于3 000,分离度不低于1.5,如图2所示。
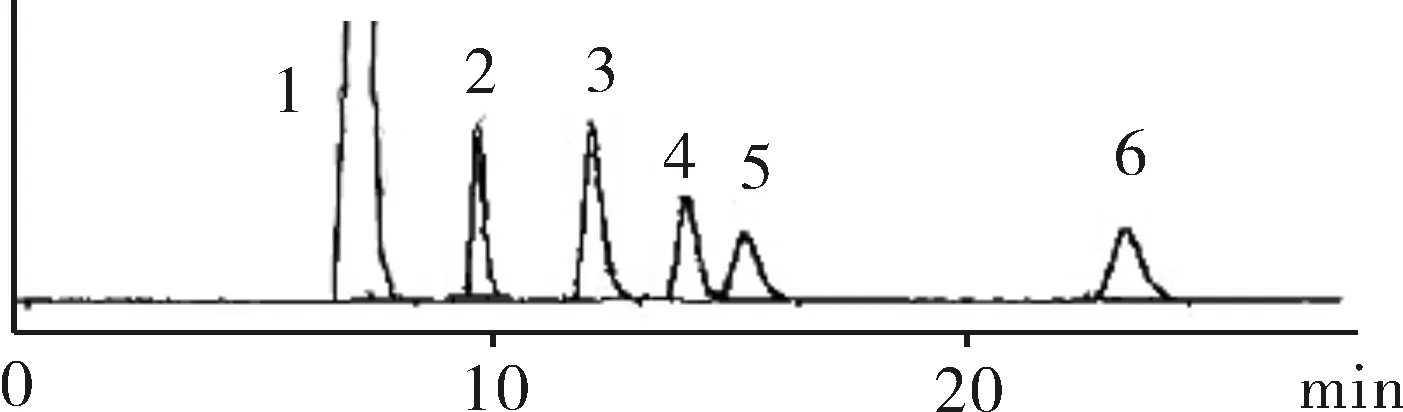
注:1.劳拉西泮;2.杂质A;3.杂质D;4.杂质E;5.杂质C;6.杂质B。
图2系统适应性试验图谱
2.3.2检测限及定量限测定取配制的各杂质对照品溶液1.0 mL,分别置于100 mL容量瓶中,加入流动相稀释至刻度,摇匀,作为检测限定量限的试验溶液, 按照上述色谱条件进样,进样10 μL,根据信噪比法进行试验,信噪比为3计算检测限,信噪比为10计算定量限,结果见表1。
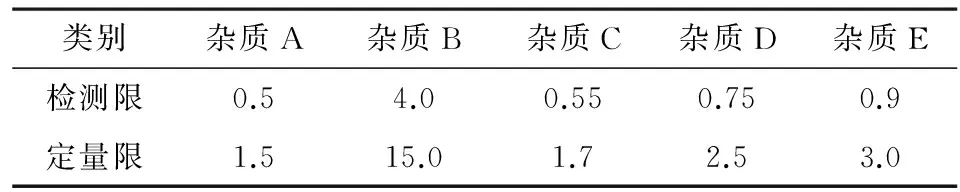
表1 各杂质检测限及定量限/ng
2.3.3线性试验精密称量各杂质对照品的贮备溶液0.5、1.0、1.5、2.0 mL,分别于50 mL容量瓶中,用流动相稀释至刻度,摇匀,作为各杂质对照品的定量溶液,分别进样10 μL,记录色谱图。以峰面积为纵坐标(A),以浓度(mg·L-1)为横坐标(C),得各杂质的回归方程及线性范围,结果见表2。
2.3.4精密度试验精密量取系统适用性溶液10 μL,重复进样6次,分别计算各峰面积的相对标准偏差。劳拉西泮和杂质A、B、C、D、E的峰面积相对标准偏差分别为0.27%、0.39%、1.04%、1.54%、0.86%、1.02%(n=6)。
2.3.5溶液稳定性试验分别精密量取系统适用性溶液10 μL,于0、2、4、8、12 h进行测定,劳拉西泮及杂质A、B、C、D、E峰面积的相对标准偏差分别为0.92%、0.83%、1.04%、0.78%、1.16%、1.28%,说明样品及各个杂质在12 h内稳定。
2.3.6回收率试验精密称取劳拉西泮对照品10.03、10.11、10.05 mg,分别置50 mL量瓶中,精密量取各杂质对照品贮备液2.5、5.0、7.5 mL,置上述量瓶中,加入流动相稀释至刻度,摇匀,分别作为加样回收率的低、中、高浓度。根据各杂质峰面积计算其回收率,结果见表3。

表3 各杂质回收率及RSD结果汇总
2.3.7专属性试验精密称定劳拉西泮对照品100 mg于50 mL的容量瓶中,加入适量甲醇振摇溶解,用甲醇稀释至刻度。量取5.0 mL对照品溶液于25 mL的容量瓶中,加入0.1 mol·L-1盐酸溶液5.0 mL摇匀,甲醇稀释至刻度,定容,置于100℃水浴锅中4 h,放冷备用。量取5.0 mL对照品溶液,加入0.1 mol·L-1氢氧化钠摇匀,甲醇稀释至刻度,定容,置于100℃水浴锅中4 h,放冷备用。量取5.0 mL对照品溶液,加入30%双氧水溶液5.0 mL,摇匀,室温放置24 h。另外分别量取5.0 mL的溶液2份,置于25 mL的容量瓶中,加入甲醇稀释至刻度。一个置于100℃水浴锅中加热4 h,放冷。另外一个置于强光(5 000 Lx)下照射12 h后,放冷。分别精密量取上述各破坏项中的溶液1.0 mL,加入流动相稀释至10.0 mL,按照上述色谱条件取10 μL进样测定,记录色谱图,见图3。

表2 各杂质线性方程及线性范围

图3 专属性试验图谱
通过图谱可以看出,劳拉西泮在碱性条件及光照条件下较为稳定,未见明显杂质出现。在酸性条件下破坏出杂质B,峰面积按照面积归一化计算约为17.6%。在氧化破坏中产生杂质C、杂质D及杂质E,按照面积归一化法计算峰面积分别为2.5%、3.1%、2.7%。高温破坏杂质主要为杂质C,峰面积按照面积归一法计算为5.1%。
2.4样品含量测定取上市品劳拉西泮片,研细,精密称量粉末适量(相当于劳拉西泮10 mg),置于50 mL容量瓶中,加入甲醇适量,超声溶解10 min,补加流动相稀释至刻度,摇匀,过滤,滤液作为供试品备用。按照上述色谱条件,精密量取10 μL进色谱仪,记录图谱。已知杂质按照杂质对照计算,未知杂质按照0.5%劳拉西泮的自身对照法来计算样品含量及有关物质含量。检查结果表明样品中杂质B和杂质E均未检出,其余有关物质和含量测定结果见表4。
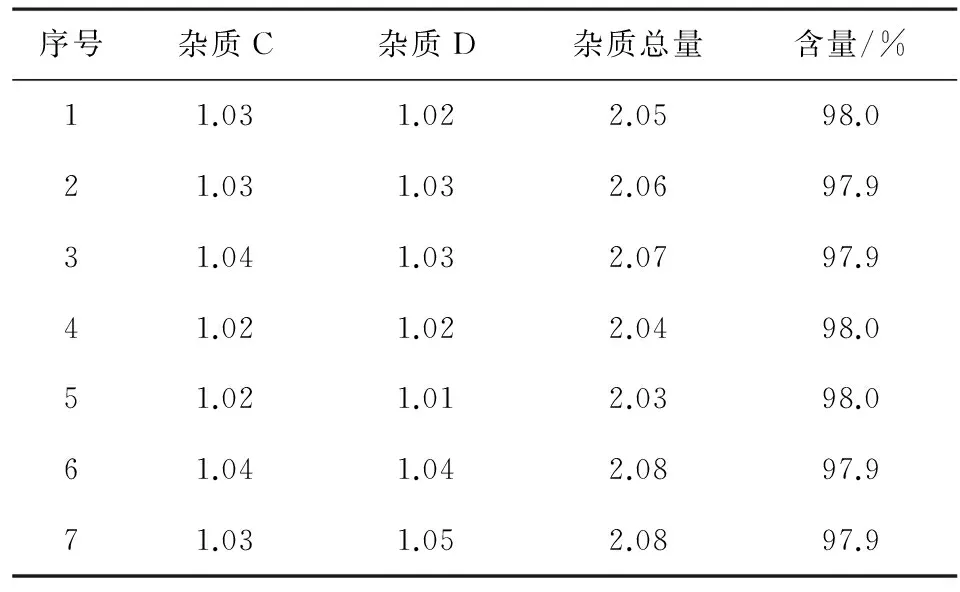
表4 劳拉西泮片有关物质及含量测定结果
3讨论
劳拉西泮在230 nm处有最大吸收,综合降解产物及各有关物质的性质,选择235 nm为有关物质及含量的检测波长,此波长可以兼顾其他各有关物质的检测。试验过程中分别试验了甲醇和磷酸缓冲盐、乙腈和磷酸缓冲盐等不同流动相,发现单一有机溶剂和磷酸缓冲盐的组合不能将劳拉西泮和各个有关物质有效分类。将甲醇和乙腈同等比例再与磷酸缓冲盐混合可以达到有效分离各个有关物质及劳拉西泮。由于杂质D为一极性较大的羧酸类化合物,保留时间受pH的影响较大。故试验中采用了磷酸盐缓冲液系统,且用磷酸调节pH为2.5。
参考文献
[1]孙定人,张石革,梁之江.国家临床新药集[M].北京:中国医药科技出版社,2001:19.
[2]郜娜,贾琳静,谢敏,等.反相高效液相色谱法测定人血浆中劳拉西泮的浓度[J].药物分析杂志,2005,25(4):423-425.
[3]程东升,陈蕾,南楠.劳拉西泮原料及其片剂中有关物质的检查[J].药物分析杂志,2006,26(12):1800-1803.
[4]The United States Pharmacopeial Convention US.Pharmacopeia (35)/ National Formulary(30)[S],2011:3720.
[5]European Directorate for the Quality Control of Medicines.European Pharmacopoeia 8.0[S],2013:1931.
[6]国家药典委员会.中国药典(二部)[S].北京:中国医药科技出版社,2015:452.
HPLC determination of the related substances in iorazepam tablets
SUN Ai-hua1,HU Wen-hui2
(1.PharmaceuticalDepartment,HuanggangCentralHospital,Huanggang,Hubei438000,China;2.EzhouCentralBloodStation,Hubei436000,China)
Abstract:ObjectiveTo determine the related substances and content in iorazepam tablets by HPLC.MethodsThe chromatographic separation was performed on ZORBAX SB-C18column(4.6 mm×250 mm,5 μm)that was made by alkyl-bonded silica gels.The mobile phase was 0.05 mol·L-1ammonium dihydrogen orthophosphate (including 0.5% trimethylamine and pH adjusted to 2.5 with phosphoric acid)∶methanol∶acetonitrile=35∶35∶30.The flow rate was 1.0 mL·min-1,the detection wavelength was 235 nm,the sample size was 10 μL,and the column temperature was set at 30℃.ResultsThe resolution between iorazepam and the other peaks met the requirements.The linearity of related substance A concentration and peak area was 1.5 μg·L-1~8.048 mg·L-1.The linearity of related substance B concentration and peak area was 15.0 μg·L-1~8.224 mg·L-1.The linearity of related substance C concentration and peak area was 1.7 μg·L-1~8.144 mg·L-1.The linearity of related substance D concentration and peak area was 2.5 μg·L-1~7.952 mg·L-1.The linearity of related substance E concentration and peak area was 3.0 μg·L-1~8.032 mg·L-1.Determination of the sample average content was 97.9%,and the related substance met the requirements.ConclusionsThe method was quick,simple,stable,and well repeatable,which could be used to determine the content of iorazepam tablets and its related substances.
Key words:iorazepam;Chromatography,High Pressure Liquid
通信作者:胡文辉,男,副主任技师,研究方向:临床检验与实验诊断,E-mail:2531134237@qq.com
doi:10.3969/j.issn.1009-6469.2016.05.014
(收稿日期:2015-10-15,修回日期:2016-02-23)
