无标记转Fat-1基因真核表达载体的构建及转基因绵羊细胞系的建立
2016-06-23阿力玛朱和平王瑞瑶闫涛苏小虎李璐王丙萍那顺温都乐齐贵春周欢敏内蒙古农业大学生命科学学院内蒙古自治区生物制造重点实验室内蒙古呼和浩特010018
阿力玛,朱和平,王瑞瑶,闫涛,苏小虎,李璐,王丙萍,那顺温都乐,齐贵春,周欢敏内蒙古农业大学 生命科学学院 内蒙古自治区生物制造重点实验室,内蒙古 呼和浩特 010018
无标记转Fat-1基因真核表达载体的构建及转基因绵羊细胞系的建立
阿力玛*,朱和平*,王瑞瑶,闫涛,苏小虎,李璐,王丙萍,那顺温都乐,齐贵春,周欢敏
内蒙古农业大学 生命科学学院 内蒙古自治区生物制造重点实验室,内蒙古呼和浩特010018
阿力玛, 朱和平, 王瑞瑶, 等. 无标记转Fat-1基因真核表达载体的构建及转基因绵羊细胞系的建立. 生物工程学报, 2016, 32(2): 212–221.
A LM, Zhu HP, Wang RY, et al. Construction of Fat-1 eukaryotic expression vector of excision markers and the establishment of transgenic sheep cell lines. Chin J Biotech, 2016, 32(2): 212–221.
摘 要:为了建立无筛选标记基因的转Fat-1基因绵羊细胞系,本研究将PCR克隆得到的Fat-1基因,合成的attB、Loxp序列并克隆入pN1-EGFP框架载体,得到可删除筛选标记基因的pEGFP-N1-Fat-1真核表达载体。体外转录合成phiC31整合酶mRNA并与线性化的pEGFP-N1-Fat-1载体共转染绵羊胎儿皮肤成纤维细胞,G418筛选得到表达绿色荧光的单克隆,再利用pET-28a-His-NLS-TAT-Cre质粒诱导Cre重组蛋白表达,将纯化后的Cre穿膜肽转导表达绿色荧光的单克隆细胞,将荧光淬灭的细胞系扩繁,提取基因组DNA,进行PCR及测序鉴定,得到无标记转Fat-1基因绵羊胎儿皮肤成纤维细胞系,为生产无筛选标记基因的转基因绵羊奠定基础。
关键词:Fat-1基因,真核表达载体,筛选标记,phiC31整合酶
Received: March 27, 2015; Accepted: November 19, 2015 Supported by: Biological High-tech Project of Inner Mongolia (No. 20030301).
*These authors contributed equally to this study.
内蒙古生物高科技项目 (No. 20030301) 资助。
1997年Spychalla等首次从秀丽隐杆线虫的基因组中获得Fat-1基因,并得到了Fat-1基因的cDNA序列,其翻译产物是ω-3多聚不饱和脂肪酸脱氢酶 (ω-3 desaturase),由402个氨基酸组成[1]。ω-3多聚不饱和脂肪酸脱氢酶以ω-6多聚不饱和脂肪酸为优先底物,使其脱氢并转化为ω-3多聚不饱和脂肪酸 (ω-3 polyunsaturated fatty acids)[2]。ω-3多聚不饱和脂肪酸为人体必需脂肪酸,具有重要的生理功能。ω-3和ω-6 PUFAS与人体健康密切相关,有研究表明ω-6/ω-3 PUFAS的比例对于维持细胞正常生长和稳态起着关键的作用。大量的流行病学资料显示,饮食中ω-6/ω-3 PUFAS比例过高,会导致机体功能障碍,引发心脑血管疾病、癌症、精神性疾病等一系列疾病的发生。但是哺乳动物自身不能合成ω-3 PUFAS,只能从深海鱼类、植物油等少数种类的外源食物中获得,随着生活水平的提高,如何补充ω-3 PUFAs已经成为人类研究的热点。转基因技术的日趋成熟使利用Fat-1转基因动物生产富含ω-3 PUFAs的农副产品,如肉制品、蛋和奶等成为可能[3],这对于提高人类身体素质有着积极的影响,也必将会带来可观的经济效益。为此,在动物中转入Fat-1基因使其在动物体内表达具有重要的研究价值[4]。
转基因技术是研究基因功能最为常用和有效的方法之一[5],转基因技术介导外源DNA整合入宿主染色体基因组上以实现基因敲除 (Knock out)、基因敲入 (Knock in) 等[6]。Cre-Loxp是目前研究最为深入的位点特异性重组系统。Cre-Loxp系统介导的重组广泛应用于哺乳动物的遗传修饰,Cre重组酶能够专一性地识别由34个碱基对的特异Loxp序列,使2个loxP之间的DNA序列发生重组[7]。如果2个Loxp位点位于一条DNA链上,且方向相同,Cre重组酶能有效切除2个Loxp位点间的序列[8]。整合的DNA多数为单拷贝外源基因,而且目的基因为定点整合。Cre-Loxp系统包括3种重组结果,这是由于Loxp位点的排列方式不同造成的:1) 2个Loxp位点在同一条DNA链上,方向相反,Cre重组酶会使2个Loxp位点间的基因序列发生倒位;2) 2个Loxp位点分别位于不同的DNA链或染色体上,Cre重组酶会使两条链间发生交换或者使染色体发生易位;3) 如果2个Loxp位点位于同一条DNA链上,方向相同,Cre重组酶就会有效切除2个Loxp位点间的序列[9]。近年来,Cre-Loxp系统被应用于小鼠、果蝇、斑马鱼、拟南芥、水稻等多种高等真核生物的遗传改造,实现外源基因在宿主染色体的定点整合或敲除、选择标记基因的删除[10]。
PhiC31整合酶来源于链霉菌噬菌体,是位点特异性重组酶丝氨酸家族的一员,可催化attB(细菌附着位点) 和attP (噬菌体附着位点) 之间的特异重组[11]。而且该反应是单向性的,重组之后生成attL和attR杂合位点,不能再成为phiC31整合酶的底物。链霉菌噬菌体phiC31整合酶能介导含attB位点的外源基因定点整合入多种真核生物基因组的假attP位点,可以维持外源基因的正常结构及高效表达[12]。但是有研究表明如果将phiC31整合酶以质粒的形式与含有attB位点的质粒进行共转染会导致宿主细胞染色体异常,可能会对宿主细胞基因组造成损伤。因此,本实验中我们利用phiC31整合酶mRNA,一种易于降解的phiC31整合酶形式介导整合事件的发生。本研究立足于ω-3 PUFAS在疾病预防与治疗和饮食平衡中的重大价值,通过基因工程的手段使Fat-1基因转入绵羊基因组中,力求生产出自身可以使ω-6/ω-3 PUFAS的比值达到平衡的转基因绵羊。另外,利用phiC31整合酶mRNA使目的基因定点整合入绵羊基因组,而最重要的是诱导表达Cre穿膜肽使设计在2个Loxp位点间的Kan+与EGFP标记基因删除,提高转基因绵羊的生物安全性,为生产无筛选标记的转基因克隆绵羊提供基础。
1 材料与方法
1.1材料
pCAGGS载体、pN1-EGFP载体与绵羊胎儿皮肤成纤维细胞由内蒙古农业大学动物生物技术重点实验室保存。pET-28a-His-NLS-TAT-Cre质粒由西北农林科技大学王凌云老师惠赠,感受态细胞BL21 (DE3) 购自北京全式金公司。
大肠杆菌DH5α菌株、小量质粒抽提试剂盒、胶回收试剂盒购自北京全式金生物技术有限公司;pMD19-T载体、限制性内切酶SalⅠ、BamHⅠ、XhoⅠ、NotⅠ、T4 DNA连接酶、LA Taq DNA聚合酶等均购自大连TaKaRa公司。引物合成、attB基因合成、基因测序均由上海生物工程公司完成。
胎牛血清、G418、NaCl、KCl、Na2HPO4等均购自Sigma公司;抗生素、酵母膏、蛋白胨和琼脂购自BBI公司。
1.2方法
1.2.1EGFP、Neo-HSV-TK-PolyA序列克隆
根据pN1-EGFP质粒上的EGFP序列设计引物,并在其上游添加Loxp序列及AgeⅠ酶切位点,在其下游引物添加NotⅠ酶切位点。引物序列见表1。
反应体系:模板pEGFP-N1 0.2 μL (837 ng/μL),EGFP上游引物2.5 μL,EGFP下游引物2.5 μL,10×缓冲液5 μL,dNTPs 3 μL,GXL DNA聚合酶0.5 μL,去离子水补至50 μL;反应条件:94 ℃5 min,94 ℃ 45 s,59 ℃ 1 min,72 ℃ 1 min,35个循环,72 ℃ 5 min。
根据pN1-EGFP质粒上的Neo-HSV-TK-PolyA序列设计引物,引物序列见表2。

表1 EGFP引物序列Table 1 The primer sequence of EGFP

表2 Neo-HSV-TK-PolyA引物序列Table 2 The primer sequence of Neo-HSV-TK-PolyA
添加Loxp序列及EcoO109Ⅰ酶切位点。反应体系与条件同1.2.1。经过胶回收,酶切连接,转化得到的载体命名为pN1-EGFP-K。
1.2.2PhiC31整合酶识别的attB序列的克隆
AttB序列送由上海生物工程公司合成,attB序列连接在pUC57载体上,并在其两端添加AseⅠ酶切位点。使用AseⅠ酶切pN1-EGFP-K及合成的attB序列,pN1-EGFP-K质粒酶切体系:10×Fast Green 缓冲液 (5 μL),pN1-EGFP-K (4 μL),Ase I酶 (1 μL),去离子水补至50 μL。经过酶切、连接与转化,载体命名为pN1-EGFP-K-attB。
1.2.3EGFP启动子CMV序列的克隆
根据pN1-EGFP质粒上的CMV序列设计引物扩增CMV片段,并在其引物上下游分别添加BamHⅠ和AgeⅠ酶切位点,引物序列见表3。
PCR反应体系:模板pN1-EGFP 0.2 μL (837 ng/μL),CMV上游引物2.5 μL,CMV下游引物2.5 μL,10×缓冲液5 μL,dNTPs 3 μL,GXL DNA聚合酶0.5 μL,去离子水补至50 μL;反应条件:94 ℃ (5 min),94 ℃ (45 s),59 ℃ (45 s),72 ℃(1 min),35个循环,72 ℃ (5 min)。经过胶回收,酶切连接,转化得到载体pN1-EGFP-K-attB-CMV。
1.2.4Fat-1基因克隆
根据GenBank上的Fat-1基因序列设计引物,在其上游添加Hind Ⅲ 酶切位点,下游添加PstⅠ酶切位点。引物序列见表4。

表3 CMV引物序列Table 3 The primer sequence of CMV

表4 Fat-1引物序列Table 4 The primer sequence of Fat-1
反应条件:98 ℃ (5 min);98 ℃ (1 min),61 ℃(3 min),72 ℃ (3 min),35个循环,72 ℃ (10 min),4 ℃ (10 min)。将PCR产物及pN1-EGFP-K-attB-CMV用Hind Ⅲ和PstⅠ双酶切后回收目的片段并连接转化,菌落PCR及酶切鉴定正确的菌株进行测序分析。重组载体命名为pEGFPN1-Fat-1 (图1)。

图1 pEGFP-N1-Fat-1质粒图谱Fig. 1 The frame of pEGFP-N1-Fat-1 plasmid.
1.2.5PhiC31整合酶mRNA体外转录
根据上海生工优化合成的phiC31整合酶序列设计引物,并在其上游添加T7启动子及BamHⅠ酶切位点,在其下游引物添加NotⅠ酶切位点。经过PCR扩增,胶回收,酶切并与pN1-EGFP载体连接,得到载体pN1-EGFP-phiC31。然后利用纯化的线性pN1-EGFP-phiC31进行体外转录,体外转录体系:5×转录反应缓冲液4 μL,ATP/CTP/GTP/UTP mix 8 μL,线性化的pN1-EGFP-phiC31 1 μL,转录酶混合物2 μL,无酶水 (DEPC) 5 μL,充分混匀,37 ℃保温2 h后对体外转录产物进行1%琼脂糖凝胶电泳检测并纯化,测定其浓度,–80 ℃保存备用。
1.2.6PhiC31整合酶mRNA与pEGFP-N1-Fat-1共转染
当传代2次的胎儿皮肤成纤维细胞生长至70%–80%时,用phiC31整合酶mRNA与pEGFP-N1-Fat-1共转染。将5 μg pEGFP-N1-Fat-1质粒及1 μg phiC31整合酶mRNA与细胞混合后加入到电击杯中,使用BTX ECM2001电穿孔仪150 V 2 ms电击一次,室温放置10 min,再将细胞转移到24孔板中加入培养液混匀放入CO2培养箱中培养,次日观察细胞生长状况,如果死细胞过多需换液,48 h后观察细胞表达绿色荧光的情况。阳性对照为转染的质粒pN1- EGFP。
1.2.7转基因细胞的DNA水平上的鉴定
经过400 μg/mL的G418培养液筛选12 d左右直至长出的阳性细胞克隆,挑取单克隆扩大培养,此时加入的G418浓度减半。细胞扩增后传至24孔板中,直到细胞传至60 mm培养皿中部分提取基因组DNA作PCR鉴定,一部分冻存。
1.2.8Cre穿膜肽切除转基因细胞中的筛选标记
用转化有pET-28a(+)-His-NLS-TAT-Cre质粒的大肠杆菌感受态细胞BL21使用IPTG诱导Cre穿膜肽表达。将细胞解冻并传代2次,与100 μg/mL的His-NLS-TAT-Cre重组蛋白共孵育4 h,蛋白转导后用PBS清洗2次,继续培养3–4 d后消化起来,将细胞稀释到很小的浓度,在显微镜下用灭过菌的吸管随机吸取单个细胞加入到96孔板的每一个孔中进行单细胞筛选,待细胞生长密度可以达到传代标准时在荧光显微镜下观察,找出无绿色荧光的单克隆做标记并扩大培养,直至培养到卡式瓶中把细胞消化起来,一部分提取基因组作鉴定,一部分冻存。
2 结果与分析
2.1EGFP、Neo-HSV-TK-PolyA序列扩增结果
根据EGFP及Neo-HSV-TK-PolyA序列的引物进行PCR扩增,扩增产物经琼脂糖凝胶电泳鉴定在771 bp及1 277 bp处有条带,与目的片段大小相符 (图2)。
pN1-EGFP-K质粒分别经过AgeⅠ、NotⅠ及StuⅠ、EcoO109Ⅰ双酶切,电泳分析显示切出大小为771 bp和1 277 bp的DNA片段 (图3)。
2.2AttB序列克隆结果
将pN1-EGFP-K-CMV-attB质粒进行AseⅠ酶切,电泳检测到3、9号载体切出300 bp左右的条带,与attB片段大小吻合 (图4),证明attB片段已经连接成功。

图2 EGFP及Neo-HSV-TK-PolyA的PCR扩增产物Fig. 2 PCR amplification products of EGFP and Neo-HSV-TK-PolyA.
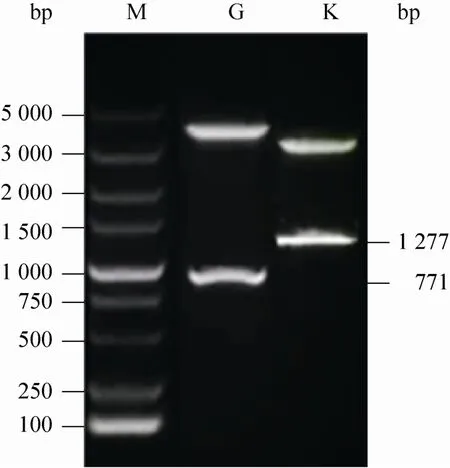
图3 pN1-EGFP-K质粒双酶切凝胶电泳结果Fig. 3 Dual-restriction digestion of pN1-EGFP-K plasmid.
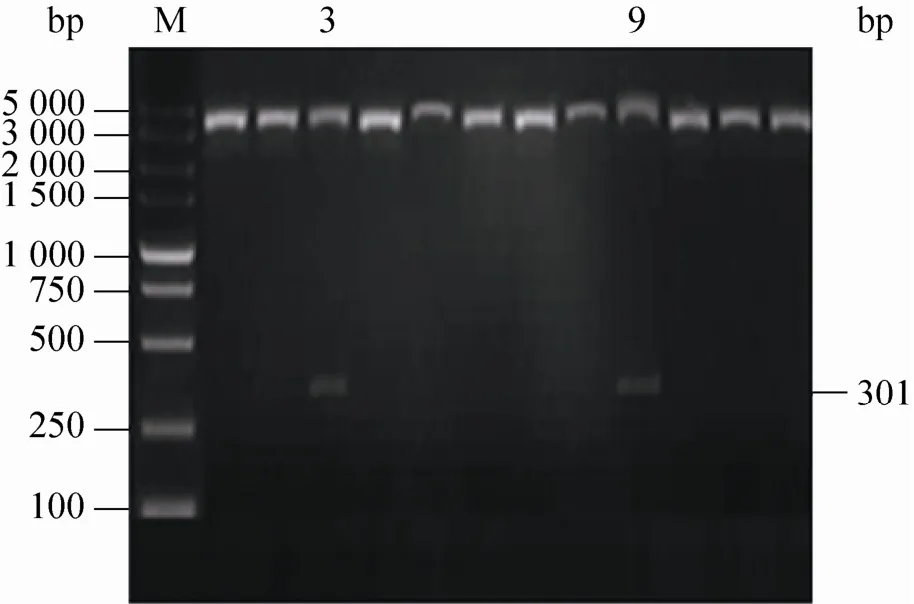
图4 pN1-EGFP-K-CMV-attB酶切鉴定Fig. 4 pN1-EGFP-K-CMV-attB restriction digestion identification result.
2.3EGFP启动子CMV序列克隆结果
2.3.1EGFP启动子CMV序列扩增
以pN1-EGFP为模板所克隆的CMV片段,经琼脂糖电泳检测约600 bp的DNA片段,与目的片段大小相符 (图5)。
2.3.2pN1-EGFP-K-attB-CMV质粒酶切鉴定结果
pN1-EGFP-K-CMV质粒经BamHⅠ和AgeⅠ双酶切,进行琼脂糖凝胶电泳,结果显示切出612 bp大小的目的条带,与预期结果相符 (图6)。
2.4Fat-1基因克隆结果
2.4.1Fat-1基因扩增结果
根据GenBank上的Fat-1基因序列,设计引物,PCR扩增,琼脂糖凝胶电泳检测出大小约1 800 bp左右的目的片段,与目的基因大小吻合 (图7)。

图5 EGFP启动子CMV扩增结果Fig. 5 EGFP promoter CMV amplification result.

图6 pN1-EGFP-K-attB-CMV质粒酶切鉴定Fig. 6 pN1-EGFP-K-attB-CMV restriction digestion identification result.

图7 Fat-1 PCR电泳结果Fig .7 Fat-1 gene electrophoresis result.
2.4.2pEGFP-N1-Fat-1重组质粒酶切鉴定结果
pEGFP-N1-Fat-1质粒经过Hind Ⅲ和PstⅠ双酶切,电泳检测到1、2、3、4号都有切下大小1 800 bp左右片段 (图8),初步验证目的基因已连接入框架载体,送往上海生物工程公司测序。测序结果与预期相同,表明pEGFP-N1-Fat-1重组载体构建成功。
2.5Fat-1基因的定点整合细胞转染
本实验以转染pN1-EGFP的细胞作为阳性对照,将phiC31整合酶mRNA与pEGFP-N1-Fat-1质粒共转染细胞,转染后48 h观察细胞转染情况。图中A、A′为转染pEGFP-N1的细胞在明场及激发光下的细胞 (lOOX),B、B′为转染phiC31整合酶mRNA与pEGFP-N1-Fat-1质粒的细胞在明场及激发光下的细胞 (lOOX),转染效率比较高 (图9)。
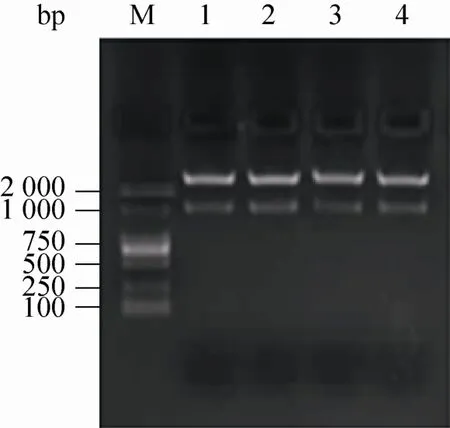
图8 pEGFP-N1-Fat-1的酶切鉴定Fig. 8 Results of enzyme digestion of recombinant plasmid of pEGFP-N1-Fat-1.

图9 细胞转染后荧光检测Fig. 9 Fluorescence detection of the transfected cells.
2.6转Fat-1基因绵羊胎儿皮肤成纤维细胞的鉴定
提取6个细胞系的基因组,对定点整合断裂位点PCR产物进行2%琼脂糖凝胶电泳,PCR检测结果显示1、2、4、5、6、8号没有条带出现 (图10),表明attB片段没有扩增出来,而对这6个基因组进行Fat-1基因PCR扩增,出现目的条带 (图11),大小与目的基因吻合,初步判定Fat-1基因已整合入绵羊基因组中。
2.7无筛选标记转Fat-1基因细胞系的鉴定
将所得到的EGFP与Fat-1基因PCR扩增产物分别进行1%琼脂糖凝胶电泳,由电泳图可见1、3、10、12号扩增出EGFP基因 (图12),而2、4、5、6、7、8、9、11、13、14号没有扩增出EGFP片段,初步断定为筛选标记已去除。取2、4、5、6、7、8、9、11、13、14号基因组进行Fat-1基因的PCR扩增发现10个样品都扩增出目的基因 (图13),初步判定这些是切除标记基因并转入Fat-1基因的细胞系。

图10 attB PCR扩增产物凝胶电泳结果Fig. 10 The results of PCR amplification products of attB.
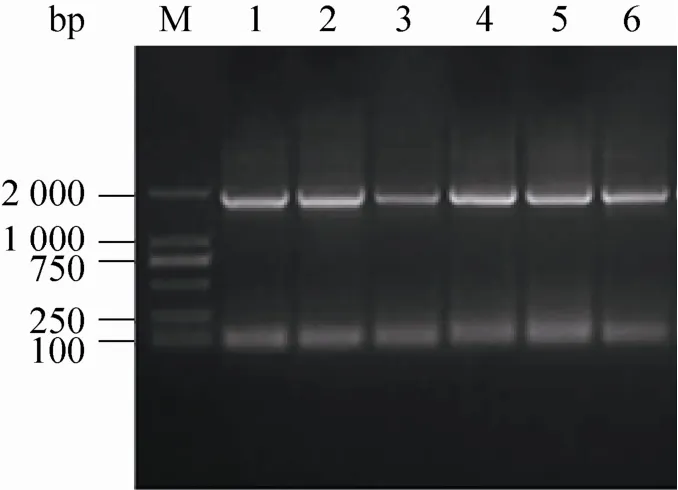
图11 Fat-1基因PCR扩增产物凝胶电泳结果Fig. 11 The results of PCR amplification products of Fat-1 gene.
3 讨论
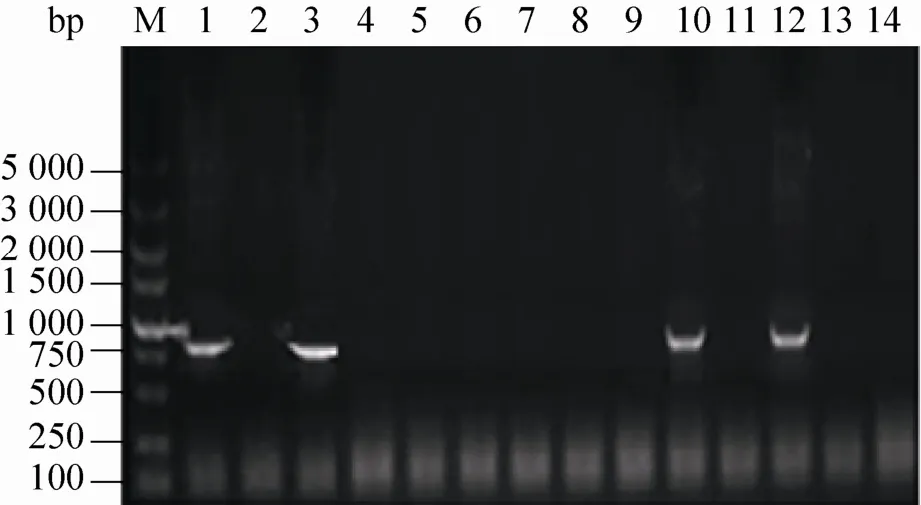
图12 EGFP基因PCR扩增产物凝胶电泳结果Fig. 12 The results of PCR amplification products of EGFP gene.

图13 Fat-1基因PCR扩增产物凝胶电泳结果Fig. 13 The results of PCR amplification products of Fat-1 gene.
多不饱和脂肪酸为人体必需脂肪酸,具有重要的生理功能。摄入高水平的ω-3PUFAS可以促进婴幼儿大脑、视网膜和神经系统的发育,还可以降低心血管疾病和炎症的发生[13]。哺乳动物体内缺乏△12以上的脂肪酸脱氢酶不能直接合成ω-3及ω-6PUFAS,只能从食物中获取[14]。因此本研究构建转入Fat-1基因的真核表达载体,为以后使其转入动物基因组中生产出可以自身合成多聚不饱和脂肪酸的转基因家畜。
PhiC31整合酶是来源于链霉菌属噬菌体的重组酶,它可以将带有attB的转基因质粒定点地整合入哺乳动物基因组假attP位点。phiC31整合酶介导的位点特异性整合较随机整合与病毒载体系统安全的多,是生产转基因动物的理想工具[15]。有研究表明,当phiC31整合酶以质粒的形式与载体共同转染细胞后,对这些细胞进行检测发现细胞核型分析异常,phiC31整合酶持续表达会影响转基因细胞的生长与后期动物的发育。因此本研究使用一种快速的易于降解的phiC31整合酶mRNA的形式与携带目的基因的质粒共转染细胞,因为mRNA易于降解,因此本实验中用到的仪器等都经过了处理,构建的含有phiC31整合酶基因的载体由上海生物工程公司合成,其功能已有研究证实比较可靠。phiC31整合酶介导的位点特异性整合远离内源基因的转录起始位点和与癌症有关的基因,这就使其避免了基因表达紊乱。因此phiC31整合酶介导的重组反应安全性比较高[16]。
自20世纪80年代以来,生物安全问题已引起世界各界的热议,2000年5月《卡塔赫纳生物安全议定书》出炉[17]。该书意在解决转基因生物安全的问题。此后,各国研究组织与机构开始关注转基因动物的安全性问题,政府也出台了相应的法律法规来管理和规范转基因动物产品的研究与应用[18]。目前,转基因食品安全在我国引发人们的热议,转基因家畜研究是一个重要的领域,因此研究降低转基因家畜安全隐患是一个关键性的问题,而转基因生产时大都会使用筛选标记基因,这些标记基因的使用和残留会引起转基因生物环境与产品的安全性[19],因此本实验中构建的载体带有两个同向的Loxp位点,而且EGFP基因与Neo-HSV-TK-PolyA基因都在两个Loxp位点之间,如果后期需要可以使构建好的载体与Cre酶共同转染删除EGFP基因和Neo-HSV-TK-PolyA基因[20]。而且本实验中构建的pEGFP-N1-Fat-1载体含有attB位点,它可以识别动物基因组上的假attP位点并在phiC31整合酶的介导下使目的基因定点整合入哺乳动物基因组中[21]。因此本文中构建的真核表达载体为无选择标记的安全转基因技术研究提供基础,特别是在将来利用本研究的结果实施选择标记的删除,从而获得无选择标记的供体细胞,通过体细胞核移植技术生产无选择标记基因的转基因个体。这将推动转基因动物育种技术向前迈出重要的一步。
REFERENCES
[1] Ruxton CH, Reed SC, Simpson MJ, et al. The health benefits of omega-3 polyunsaturated fatty acids: a review of the evidence. J Hum Nutr Diet, 2004, 17(5): 449–459.
[2] Wu X, Ouyang HS, Duan B, et al. Production of cloned transgenic cow expressing omega-3 fatty acids. Transgenic Res, 2012, 21(3): 537–543.
[3] Yao QH, Zhang XC, Fu T, et al. ω-3 polyunsaturated fatty acids inhibit the proliferation of the lung adenocarcinoma cell line A549 in vitro. Mol Med Rep, 2014, 9(2): 401–406.
[4] De Carlo F, Witte TR, Hardman WE, et al. Omega-3 eicosapentaenoic acid decreases CD133 colon cancer stem-like cell marker expression while increasing sensitivity to chemotherapy. PLoS ONE, 2013, 8(7): e69760.
[5] Powell BC, Walker SK, Bawden CS, et al. Transgenic sheep and wool growth: possibilitiesand current status. Reprod Fertil Dev, 1994, 6(5): 615–623.
[6] Lan C, Ren LN, Wu M, et al. Deletion of marker gene in transgenic goat by Cre/LoxP system. Chin J Biotech, 2013, 29(12): 1847–1854 (in Chinese).
兰翀, 任丽娜, 吴敏, 等. 利用Cre/LoxP系统删除转基因山羊体内的选择标记基因. 生物工程学报, 2013, 29(12): 1847–1854.
[7] Zhao Y, Yu T, Xing SC. Application of Cre/lox site-specific recombination system in transgenic plants. Chin J Biochem Mol Biol, 2010, 26(2): 95–103 (in Chinese).
赵妍, 余涛, 邢少辰. Cre/lox位点特异重组系统在转基因植物中的应用. 中国生物化学与分子生物学报, 2010, 26(2) : 95–103.
[8] Wang ZR, Liu XM, Zhou JR, et al. Efficacy evaluation of Cre-LoxP recombinase system to delete the endogenous selectable marker gene. Chin J Biochem Mol Biol, 2014, 30(2): 194–201 (in Chinese).
王志蕊, 刘西梅, 周荆荣, 等. Cre-Loxp重组系统删除内源性选择标记基因的效能评价. 中国生物化学与分子生物学报, 2014, 30(2): 194–201.
[9] Lu JJ, Maddison LA, Chen WB. φC31 integrase induces efficient site-specific excision in zebrafish. Transg Res, 2011, 20(1): 183–189.
[10] Li L, Guo R, Chang XS, et al. Generation of islet β cell-specific Cre recombinase targeting vector by homologous recombination in bacteria. Acad J Sec Milit Med Univ, 2014, 35(2): 185–190 (in Chinese).
李玲, 国蓉, 常绪生, 等. 利用细菌内同源重组技术构建胰岛β细胞特异性表达Cre重组酶的基因敲入打靶载体. 第二军医大学学报, 2014, 35(2): 185–190.
[11] Kempe K, Rubtsova M, Berger C, et al. Transgene excision from wheat chromosomes by phage φC31 integrase. Plant Mol Biol, 2010, 72(6): 673–687.
[12] Bi YX, Liu XM, Hua ZD, et al. Site-specific modification of pig genome mediated by ΦC31 integrase. Chin J Biochem Mol Biol, 2014, 30(2): 187–193 (in Chinese).
毕延震, 刘西梅, 华再东, 等. ΦC31整合酶介导猪基因组定点修饰的探讨. 中国生物化学与分子生物学报, 2014, 30(2): 187–193.
[13] Benbrook CM, Butler G, Latif MA, et al. Organic production enhances milk nutritional quality by shifting fatty acid composition: a United States-wide, 18-month study. PLoS ONE, 2013, 8(12): e82429.
[14] Lister JA. Transgene excision in zebrafish using the φC31 integrase. Genesis, 2010, 48(2): 137–143.
[15] Allen BG, Weeks DL. Transgenic Xenopus laevis embryos can be generated using φC31 integrase. Nat Methods, 2005, 2(12): 975–979.
[16] Zou LT. Study the gene inversion and deletion mediated by phage phiC31/att site-pecific recombinant system[D]. Chongqing: Southwest University, 2014 (in Chinese).
邹丽婷. 噬菌体phiC31/att位点重组系统介导的基因倒位和删除研究[D]. 重庆: 西南大学, 2014.
[17] Wang ZP, Zhang SZ. Safety of transgenic animal food. J Anhui Agri Sci, 2007, 35(19): 5868–5869 (in Chinese).
王正鹏, 张树珍. 转基因动物食品的安全性. 安徽农业科学, 2007, 35(19): 5868–5869.
[18] Wang L, Li XL. Ethical dilemmas of safety of genetically modified food in China. J Anhui Agri Sci, 2014, 42(33): 1854–1856 (in Chinese).
王磊, 李晓兰. 我国转基因食品安全的伦理困境.安徽农业科学, 2014, 42(33): 11854–11856.
[19] Pandey SN, Lee YC, Yokota T, et al. Morpholino treatment improves muscle function and pathology of Pitx1 transgenic mice. Mol Ther, 2014, 22(2): 390–396.
[20] Smith MC, Brown WR, McEwan AR, et al. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem Soc Trans, 2010, 38(2): 388–394.
[21] Chavez CL, Calos MP. Therapeutic applications of the PhiC31 integrase system. Curr Gene Ther, 2011, 11(5): 375–381.
(本文责编陈宏宇)
Construction of Fat-1 eukaryotic expression vector of excision markers and the establishment of transgenic sheep cell lines
Lima A*, Heping Zhu*, Ruiyao Wang, Tao Yan, Xiaohu Su, Lu Li, Bingping Wang, Shunwendoule Na, Guichun Qi, and Huanmin Zhou
Key Laboratory of Biological Manufacturing of Inner Mongolia Autonomous Region College of Life Science, Inner Mongolia Agricultural University, Hohhot 010018, Inner Mongolia, China
Abstract:In order to establish marker-free transgenic cell lines, we cloned Fat-1 gene, attB and Loxp sequences by PCR.Then we inserted these sequences to pN1-EGFP vector and got pEGFP-N1-Fat-1 expression vector. PhiC31 integrase mRNA which was generated by in vitro transcription and a pEGFP-N1-Fat-1 expression vector co-electroporated into sheep fetal fibroblasts, and then we got transgenic cell lines expressing green fluorescence. Prokaryotic expression and purification of Cre recombinant protein was performed. Cre recombinant protein was transducted into stably-transfected cell colonies. We identified cell colonies by sequencing and established marker-free transgenic cell lines and eventually established marker-free transgenic cell lines which were building more safely basic for producing Fat-1 transgenic animals.
Keywords:Fat-1 gene, eukaryotic expression vector, marker gene, phiC31 integrase
Corresponding author:Huanmin Zhou. E-mail: huanminzhou@126.com
