麦冬中萜类成分龙脑次苷的合成研究
2016-06-17程冬萍周侠毅宋立芳
程冬萍,周侠毅,宋立芳
(浙江工业大学 药学院,浙江 杭州 310014)
麦冬中萜类成分龙脑次苷的合成研究
程冬萍,周侠毅,宋立芳
(浙江工业大学 药学院,浙江 杭州 310014)
摘要:采用2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖与三氯乙腈亲核加成得到2,3,4,6-四-O-乙酰基-α-D-吡喃葡萄糖三氯乙酰亚胺酯,亚胺酯与l-龙脑进行糖苷化,再用甲醇钠脱保护得到龙脑次苷。并着重对糖苷化一步的反应条件进行探索优化,得到了较优的反应条件:当投料比为n(亚胺酯)∶n(l-龙脑)=1.3,催化剂三氟化硼·乙醚用量为10%,二氯甲烷中-15 ℃反应5 h,收率65.0%.
关键词:龙脑次苷;三氯乙酰亚胺酯;糖苷化;脱保护
龙脑次苷是自然界中稀有的双环单萜葡萄糖苷,是中药麦冬的特征性成分之一[1].由于龙脑次苷在麦冬中的含量低微,如果采用传统的提取分离方法从中药麦冬来获得龙脑次苷,则生产成本高昂.文献[2]报道龙脑次苷的合成方法是以四乙酰基溴代葡萄糖作为糖基供体,在碳酸银的催化下,和苷元l-龙脑进行糖苷化反应,再经过脱保护得到龙脑次苷,报道收率为35%.但是此方法不仅使用了价格昂贵的重金属盐催化剂,而且底物溴代葡萄糖不稳定,反应总收率低下.因此,改用三氯乙酰亚胺酯作为糖元供体,与传统的溴代法相比,收率高,操作简便.其中重点研究了葡萄糖三氯乙酰亚胺酯与l-龙脑的糖苷化条件.
1合成工艺探索及优化
2,3,4,6-四-O-乙酰基-α-D-吡喃葡萄糖三氯乙酰亚胺酯Ⅳ的合成路线为
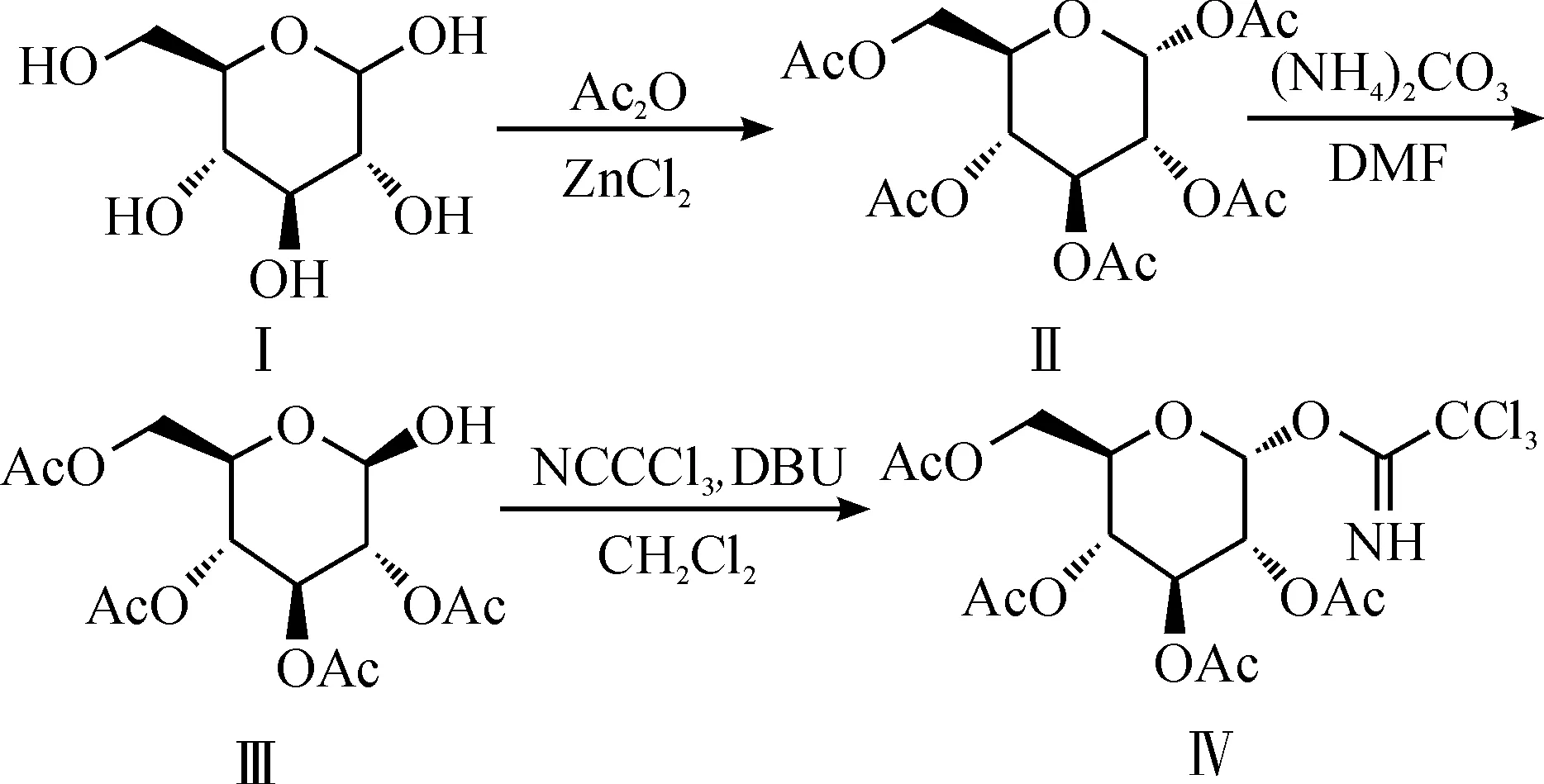
其中第一步乙酰化据文献报道有醋酸酐-醋酸钠法[3]、硫酸-醋酸酐法[4]、氯化锌-醋酐法[5]和三氯化铁-醋酐法[6]等,这几种方法收率都在70%~80%之间.醋酸酐-醋酸钠法和三氯化铁-醋酸酐法都要消耗大量的醋酐,成本高,后处理难度大;硫酸-醋酐法以浓硫酸做催化剂,浓硫酸酸性强,反应剧烈,有一定危险性;氯化锌-醋酐法以氯化锌为催化剂,作用较温和.该反应D-葡萄糖Ⅰ与溶剂醋酐的投料比为1∶6 g/mL,氯化锌与Ⅰ用量的摩尔比为40%,收率可达85%.
第二步脱1位乙酰基应有两种方法,一是与溴化氢、碳酸银反应[7],二是与碳酸铵反应[8].前者操作较繁琐,总收率较低,还使用了毒性大的溴化氢气体和高成本的银盐.相比之下,碳酸铵法应用更广泛.其中底物五乙酰基葡萄糖Ⅱ与溶剂DMF的投料比为1∶2 g/mL,碳酸铵与Ⅱ用量的摩尔比为2,收率可达90%.
第三步亚胺化多采用1, 8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)[9]催化,反应条件温和,收率高,后处理简单.催化剂DBU的量一般在10%以内即可,实验中投量为7%,三氯乙腈与底物用量的摩尔比为2,收率可达65%.
故最终选用以上合成路线制备化合物Ⅳ.
l-龙脑2-O-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷Ⅴ的合成路线为
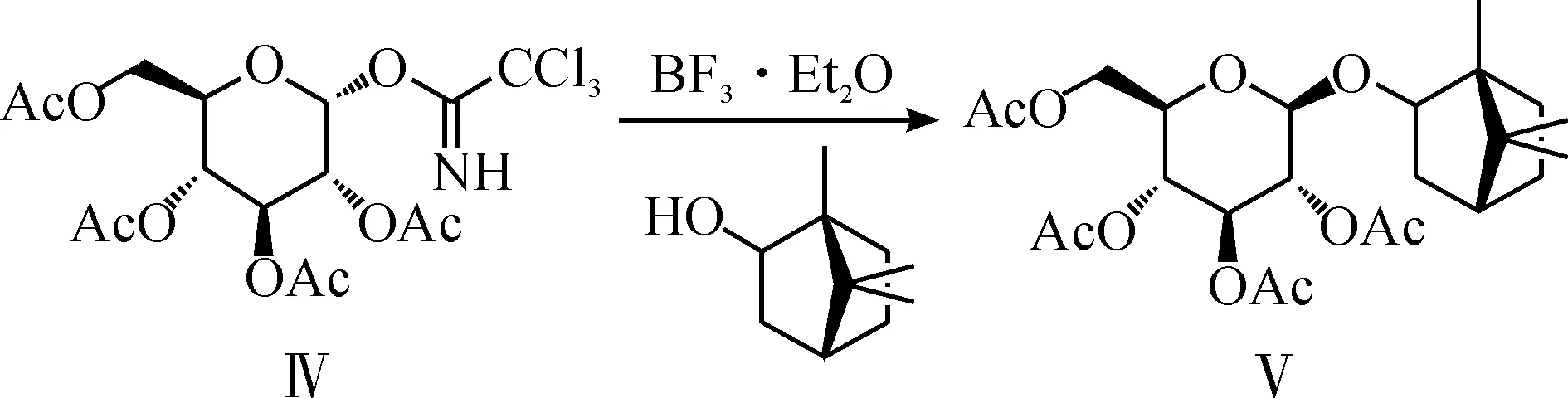
该反应一般要路易斯酸作催化剂,如三氟甲磺酸三甲基硅酯[10]、三氟化硼·乙醚[11]和对甲苯磺酸[12]等.
1.1催化剂选择
本研究考察了四种催化剂,三氟化硼·乙醚、三氟甲磺酸酐、苯磺酸以及三氟甲磺酸三甲基硅酯.将1 mmoll-龙脑和1.3 mmol三氯乙酰亚胺酯溶于5 mL二氯甲烷,氮气保护,于5 ℃下加入1.0 mmol催化剂,搅拌过夜,反应结果如表1所示.

表1 催化剂筛选
由实验结果看出:以三氟甲磺酸酐催化无目标产物生成,以苯磺酸催化有极少量产物,以三氟甲磺酸三甲基硅酯催化生成的是α构型的产物,只有以三氟化硼·乙醚催化时可得到约40%的目标产物.初步确定三氟化硼·乙醚作为糖苷化反应的催化剂.
1.2单因素试验
根据上述结果,投入1 mmoll-龙脑,1.3 mmol三氯乙酰亚胺酯,5 mL二氯甲烷和三氟化硼·乙醚,5 ℃反应过夜.结果见表2.
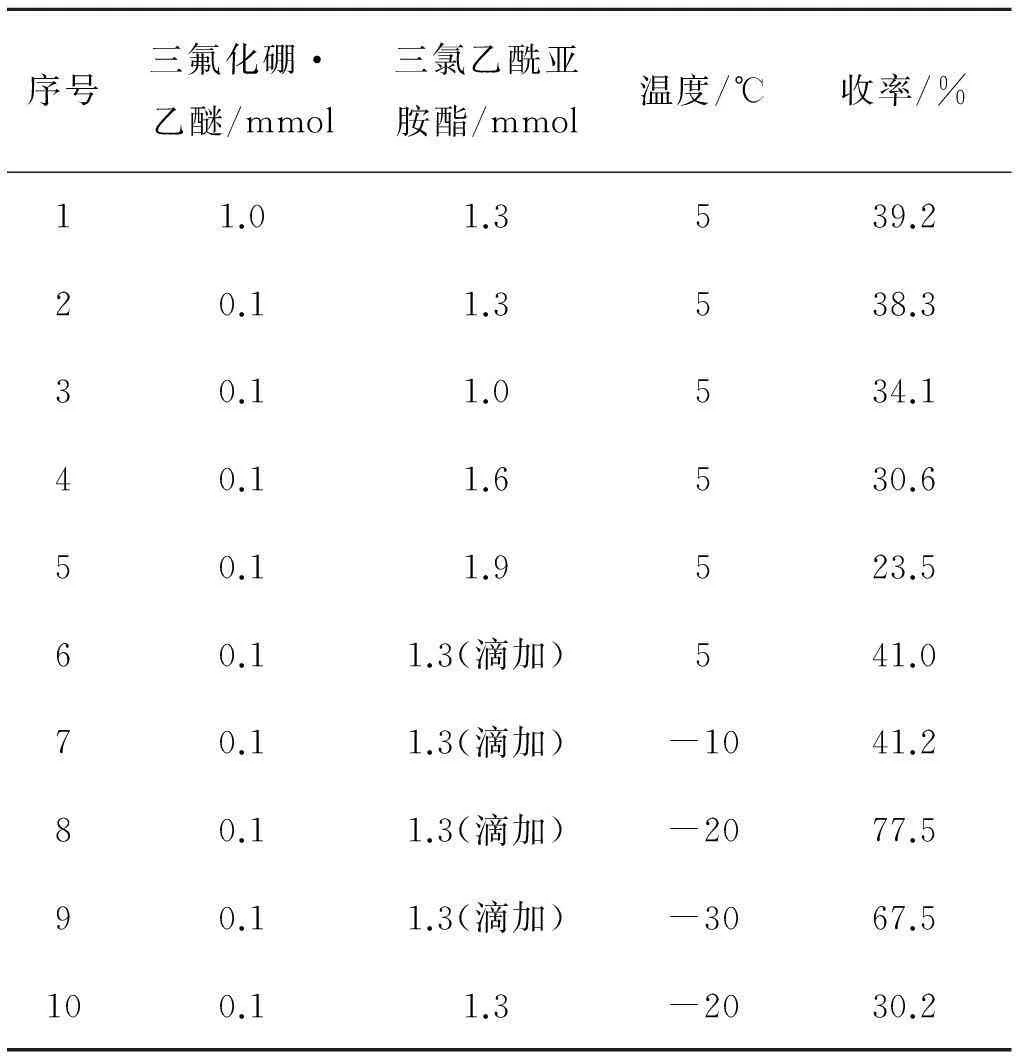
表2 单因素试验表1)
注:1) 表中底物l-龙脑用量均为1 mmol,所有反应均在氮气保护下进行.
由表2中可以得出:当三氟化硼·乙醚降至0.1 mmol,收率仍能达到38%,与1.0 mmol的收率相比无明显变化,因此三氟化硼·乙醚用量为0.1 mmol即可.
根据三氟化硼·乙醚的考察结果,投入1 mmoll-龙脑,0.1 mmol三氟化硼·乙醚,5 mL二氯甲烷,5 ℃反应,考察三氯乙酰亚胺酯的用量对反应的影响(表2中第二至五组).可以得出三氯乙酰亚胺酯的量由1.0 mmol增至1.3 mmol,收率有所提高,但继续增加三氯乙酰亚胺酯,收率开始降低.选取其中收率最好的1.3 mmol,滴加三氯乙酰亚胺酯(二氯甲烷溶解),收率略有提高(表2中第二组与第六组).故三氯乙酰亚胺酯暂且选择滴加,用量为1.3 mmol.
确定投料比、催化剂用量以及加料方式后,投入1 mmol的l-龙脑,5 mL的二氯甲烷,1.3 mmol的三氯乙酰亚胺酯,0.1 mmol的三氟化硼·乙醚,其中4 mL二氯甲烷用于溶解龙脑,剩余1 mL二氯甲烷用于溶解三氯乙酰亚胺酯且滴加,考察了温度对反应的影响.
总体来看,-20 ℃左右是较适宜的反应温度.为验证三氯乙酰亚胺酯的加入方式对收率的影响,特地补做一组-20 ℃的实验,直接加入三氯乙酰亚胺酯而非滴加,收率仅为30.2%(该条件下反应收率较5 ℃低,其可能原因为在低温下体系中水分含量较高,三氯乙酰亚胺酯水解更严重),故必须滴加.
确定最佳反应条件后,考察了反应时间的影响,通过TLC检测反应进度后发现,反应5 h后原料l-龙脑以完全消失,故选择反应时间为5 h.
1.3正交试验分析及稳定性考察
根据以上的单因素试验确定了三氟化硼·乙醚、三氯乙酰亚胺酯和温度三个因素,每个因素设三个水平,如表3所示.L9(33)正交实验结果见表4.
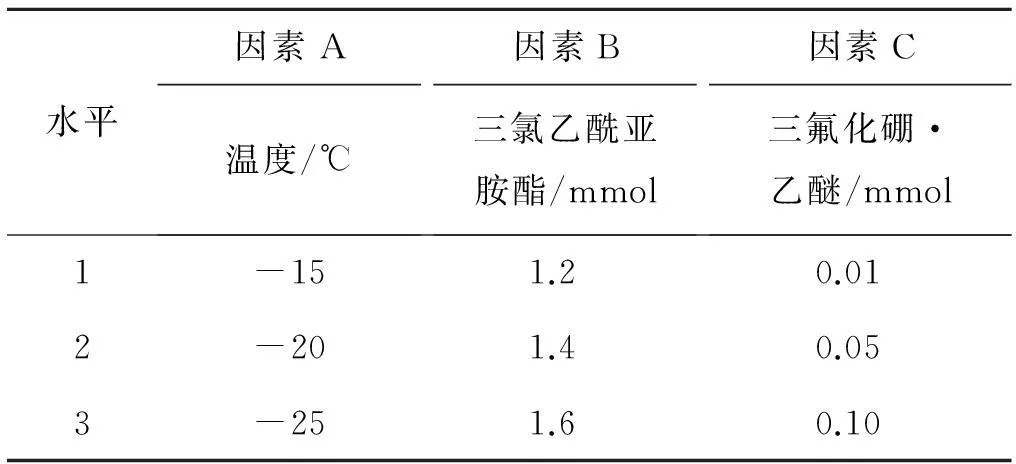
表3 正交试验的因素和水平1)
注:1) 表中底物l-龙脑用量均为1 mmol,所有反应均在氮气保护下进行,反应时间为5 h.
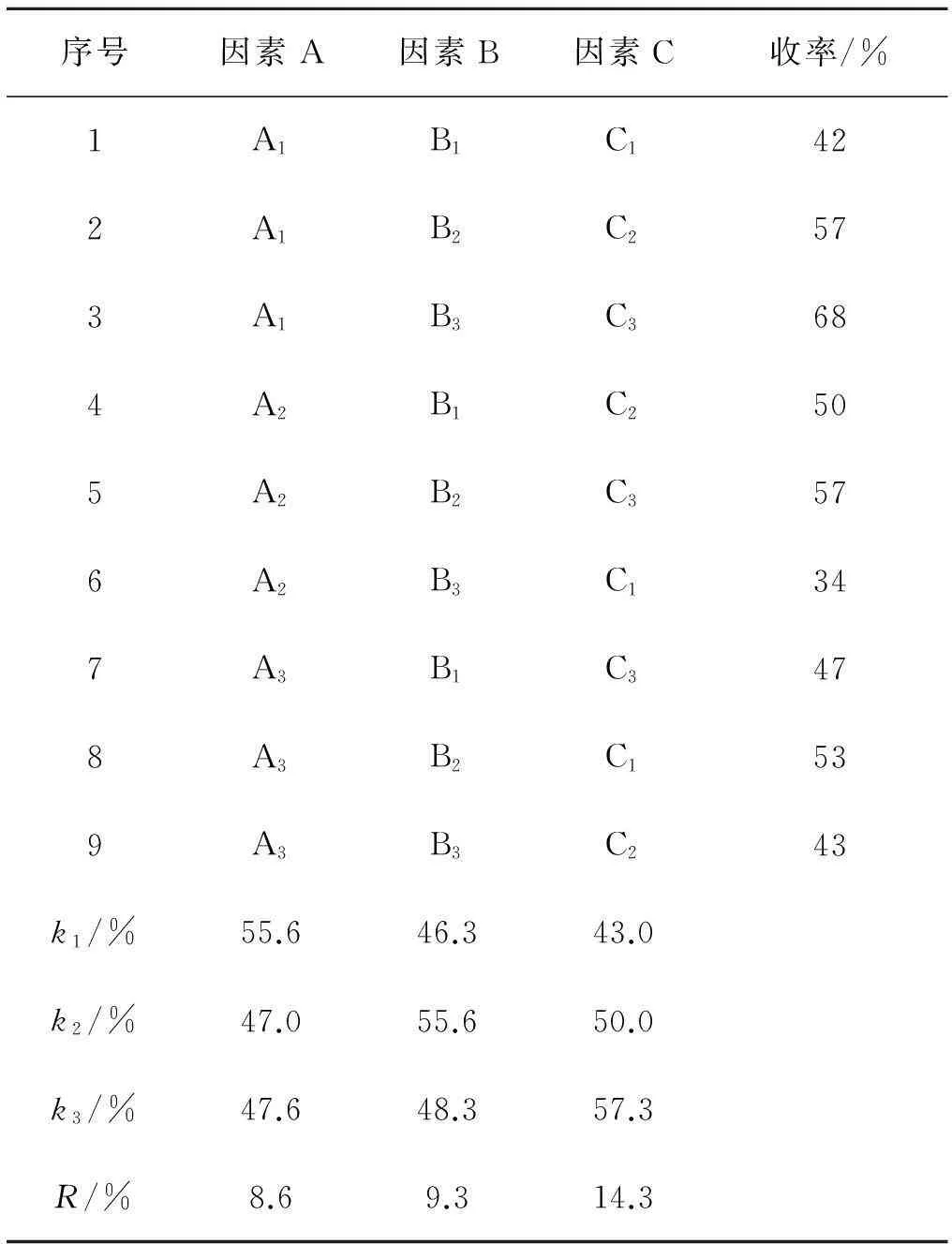
表4 L9(33)糖苷化反应正交设计实验结果
观察表4的极差分析,影响反应收率的最大因素是三氟化硼·乙醚的量,其次是三氯乙酰亚胺酯,温度对收率影响较小.温度的k1最大,亚胺酯k2最大,三氟化硼·乙醚k3最大,因此正交实验所得的最佳反应条件组合为A1B2C3,即温度为-15 ℃,三氯乙酰亚胺酯是1.4 mmol,三氟化硼·乙醚的量是0.1 mmol.
观察表4中各因素在不同水平下的收率变化,反应温度从-15 ℃降至-20 ℃,收率明显下降,由-20 ℃降至-25 ℃,收率基本平稳.因此在考查范围内,-15 ℃反应效果较好.温度-15 ℃继续升高,反应收率仍有提高的可能,故补做一组实验,三氯乙酰亚胺酯的用量为1.4 mmol,催化剂为0.1 mmol,-10 ℃反应,收率为66.0%,因此以-15 ℃为最佳温度.
三氯乙酰亚胺酯为1.4 mmol时收率有最大值,故以1.4 mmol为最佳投量.
三氟化硼·乙醚从0.01 mmol增加至0.05 mmol,再增加至0.1 mmol,收率几乎呈直线式增长.虽然前述单因素实验中0.1 mmol与1.0 mmol的收率无明显差异,但不能确定继续增加三氟化硼·乙醚是否会使收率提高.因此补做一组实验,1.4 mmol三氯乙酰亚胺酯,三氟化硼·乙醚增至0.2 mmol,-15 ℃反应,收率79.1%,与0.1 mmol时无明显变化,故选0.1 mmol为最佳投量.
综上所述,最佳组合为A1B2C3,即-15 ℃,三氯乙酰亚胺酯为1.4 mmol,催化剂为0.1 mmol.对该组条件做稳定性实验,收率与前述实验结果基本吻合,如表5所示.
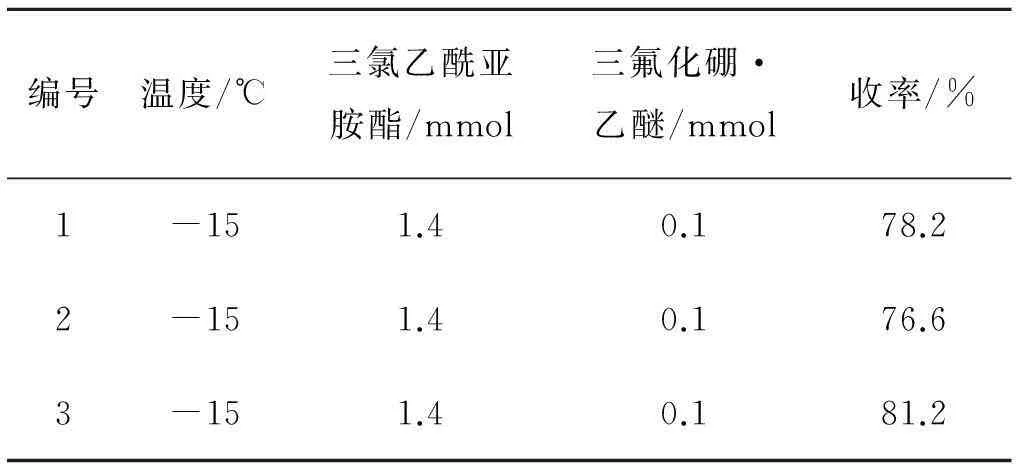
表5 稳定性实验1)
注:1) 表中底物l-龙脑用量均为1 mmol,所有反应均在氮气保护下进行,反应时间为5 h.
2实验部分
2.1主要仪器与试剂
主要仪器:X-4 型数字显示显微熔点测定仪(温度未校正);Bruker Avance III(500M)核磁共振仪.
主要试剂:D-葡萄糖(CP,国药集团化学试剂有限公司) ;醋酐(AR,杭州双林化工试剂厂) ;三氯乙腈(AR,无锡海硕生物有限公司) ;DBU (AR,杭州双林化工试剂厂) ;三氟化硼·乙醚(AR,阿拉丁试剂公司) ;l-龙脑(AR,阿拉丁试剂公司).
2.2实验过程与方法
龙脑次苷的总合成路线为
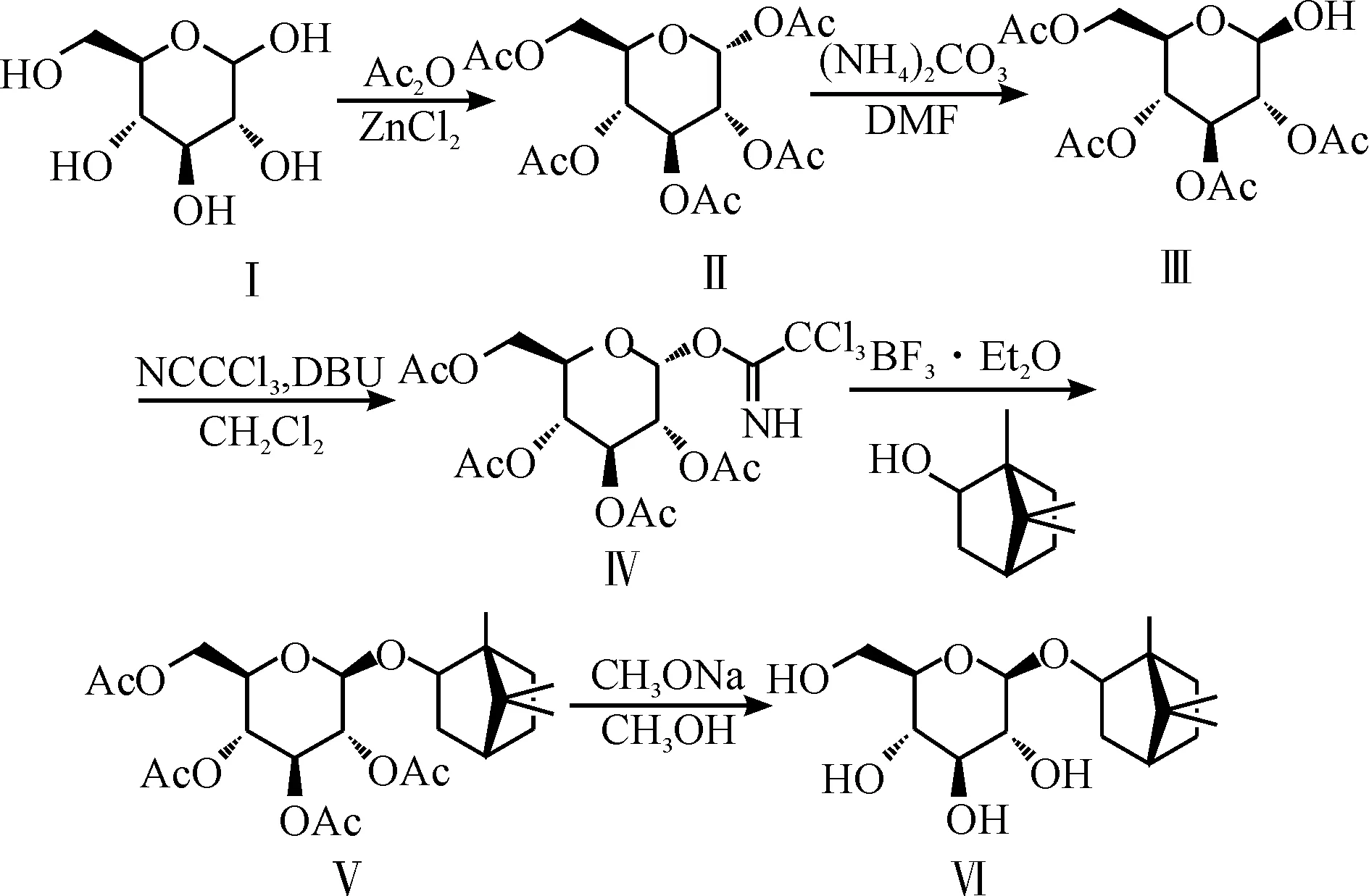
1,2,3,4,6-五-O-乙酰基-α-D-吡喃葡萄糖Ⅱ:在1 000 mL圆底烧瓶中加入600 mL醋酸酐和30.2 g(0.222 mol)氯化锌[5],加热至60 ℃使氯化锌溶解.停止加热,在60 ℃以下分批加入100 g D-葡萄糖Ⅰ,约45 min加完.然后100 ℃回流4 h,降至室温,减压蒸馏除去未反应的醋酸酐,将剩余溶液倒入3 000 mL冰水中,搅拌约2 h待晶体充分析出后,过滤,洗涤,用50%乙醇水溶液重结晶,60 ℃减压干燥得白色粉末162.10 g,收率82.4%,m.p.110~111 ℃.
2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖Ⅲ:称取97.6 g (0.25 mol)Ⅱ,2.48 g (0.5 mol)碳酸铵,加入到195 mL DMF中,40 ℃反应6 h[8],用乙酸乙酯萃取,有机相用饱和食盐水洗涤,无水硫酸钠干燥,减压除去溶剂得80.4 g淡黄色糖浆,收率92.4%.
2,3,4,6-四-O-乙酰基-α-D-吡喃葡萄糖三氯乙酰亚胺酯Ⅳ:将174.2 g (0.5 mol) Ⅲ和144.4 g(1.0 mol)三氯乙腈加入到350 mL干燥的二氯甲烷中溶解,降至0 ℃,滴加入5.2 mL (0.035 mmol) DBU,室温反应过夜[9].次日旋干溶剂,柱层析分离得160.1 g淡黄色糖浆,收率65.0%.1H-NMR (500 MHz, CDCl3): δ 1.97(s, 3H), 1.98(s, 3H), 2.00(s, 3H), 2.02(s, 3H), 4.07(dd,J=12.4 Hz, 1.7Hz, 1H), 4.17(m, 1H), 4.23(dd,J=12.4, 4.13 Hz, 1H), 5.09(dd,J=10.1, 3.61 Hz, 1H), 5.13(t,J=9.8 Hz, 1H), 5.51(t,J=9.8 Hz, 1H), 6.51(d,J=3.7 Hz, 1H), 9.69(s, 1H).

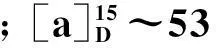
3结论
以廉价易得的D-葡萄糖为原料经过全乙酰化、脱1位乙酰基、与三氯乙腈亲核加成后,再与l-龙脑进行糖苷化,最后通过甲醇钠脱保护即为龙脑次苷,共5步反应,总收率35.4%.每步反应均具有良好的收率,且反应过程及后处理均较为简便.其中糖苷化是比较困难的一步.对于该步反应我们摈弃了传统的溴代糖法,用三氯乙酰亚胺酯法借助邻基参与效应高选择性地合成了龙脑次苷.并对其进行单因素考察和正交试验优化,最后将该步反应收率提升至79%.三氯乙酰亚胺酯法成功用于合成龙脑次苷,为其它单萜葡萄糖苷尤其是大位阻的双环单萜葡萄糖苷的合成提供了思路.
参考文献:
[1]程志红,李林洲,刘楠等.中药麦冬脂溶性化学成分的研究[J].中国药学杂志,2005,40(5):337-341.
[2]PATOV S A, PUNEGOV V V. Synthesis of certain monoterpenoid glucosides[J]. Chemistry of natural compounds,2006,42(4):431-433.
[3]DEREDAS D, FRANKOWSKI A. Synthesis of d-erythrofuranosyl C-nucleosides of imidazole from 4(5)-(D-arabino- tetritol-1-yl) imidazole[J]. Carbohydrate research,1994,252:275-281.
[4]LICHTENTHALER F W, KOEHLER B. An expedient route to acylated glucosyl halides with a free 2-OH group[J]. Carbohydrate research,1994,258(1/2):77-85.
[5]俞继华,肖红新.α-五酰葡萄糖的合成[J].化学推进剂与高分子材料,2000(3):28-28.
[6]刘媛媛,刘伯成.五乙酰葡萄糖的合成工艺研究[J].科技视界,2013(29):422-423.
[7]于小凤,孙玉.2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖的合成工艺改进[J].化工进展,2010,29(2):334-337.
[8]DORST J A, HEUSDEN ] C J, TIKKANEN J M. Synthesis of Hexp-(1→4)-β-D-GlcpNAc-(1→2)-α-D-Manp-(1→O)(CH2)7CH3probes for exploration of the substrate specificity of glycosyltransferases: part II Hex=3-O-methyl-β-D-Gal, 3-deoxy-β-D-Gal, 3-deoxy-3- fluoro-β-D-Gal, 3-amino-3-deoxy-β-D-Gal,β-D-Gul,α-L-Alt, orβ-L-Gal[J]. Carbohydrate research,1997,297(3):209-227.
[9]傅大双,张首国,彭涛,等.合成2,3,4,6-O-四苄基-α-D-葡萄糖三氯乙酰亚胺酯的工艺改进[J].合成化学,2013,21(3):333-335.
[10]周峰岩.苯丙素苷类化合物的合成[D].杭州:浙江大学,2005.
[11]LI W, WU X F, TONG Y F,; et al. Total synthesis of adicardin[J]. Journal of Asian natural products research,2009,11(8):720-727.
[12]HANAURA M, AGOCS A. New methods for the synthesis of carotenoid glycosides[J]. Tetrahedron letters,2014,55(26):3625-3627.
(责任编辑:陈石平)
A study on the synthesis ofl-borneol 2-O-β-D-glucopyranoside in radix ophiopogonis
CHENG Dongping, ZHOU Xiayi, SONG Lifang
(College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China)
Abstract:2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl trichloroacetimidate was obtained by 2,3,4,6-tetra-O-acetyl-β-D-glucose reacting with trichloroacetonitrile, and then reacted with l-borneol to give l-borneol -2-O-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside through glycosidation. The target product l-borneol 2-O-β-D-glucopyranoside was formed through the deprotection of l-borneol -2-O-2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside in methanol with sodium methoxide as base. The reaction conditions were optimized and the optimal process conditions were obtained. l-borneol reacted with trichloroacetimidate (1.3 equiv.) in dichloromethane at -15 ℃ for 5 h in the presence of 0.1 equivalents of boron fluoride ethyl ether to give an optimum yield of 65.0%.
Keywords:l-borneol-2-O-β-D-glucopyranoside; trichloroacetic imine esters; glycosylation; deprotection
收稿日期:2015-09-30
作者简介:程冬萍(1976—),女,浙江温州人,副教授,博士,研究方向药物和药物中间体的合成研究,E-mail:chengdp@zjut.edu.cn.
中图分类号:O629.13
文献标志码:A
文章编号:1006-4303(2016)02-0169-05
