HEK293细胞中杂合状态下无义突变体L539fs/47对野生型HERG电流的作用
2016-06-06张军波张爱峰刘仲伟王军奎韩稳琦潘军强李国良孙超峰
吕 颖,张军波,张爱峰,刘仲伟,王军奎,韩稳琦,潘军强,李国良,孙超峰
(1. 陕西省人民医院心内一科,陕西西安 710068;2. 西安交通大学第一附属医院心内科,陕西西安 710061;3. 西安交通大学第二附属医院心内科,陕西西安 710004)
◇基础研究◇
HEK293细胞中杂合状态下无义突变体L539fs/47对野生型HERG电流的作用
吕颖1,张军波2,张爱峰3,刘仲伟1,王军奎1,韩稳琦2,潘军强1,李国良2,孙超峰2
(1. 陕西省人民医院心内一科,陕西西安710068;2. 西安交通大学第一附属医院心内科,陕西西安710061;3. 西安交通大学第二附属医院心内科,陕西西安710004)
摘要:目的研究单独表达无功能的无义突变体L539fs/47在HEK293细胞中杂合状态下对野生型HERG电流的作用。方法用脂质体转染法将野生型HERG及突变体HERG-L539fs/47分别转染HEK293细胞36 h后,RT-PCR检测HERG mRNA表达,免疫荧光及免疫印迹检测HERG蛋白定位及表达量,全细胞膜片钳检测IHERG电流密度。结果野生组HERG的mRNA表达量高于L539fs/47突变组(1.066±0.612 vs. 0.254±0.397, P<0.01)。免疫荧光检测发现野生型HERG蛋白多于突变体,野生型HERG蛋白主要分布在细胞膜上;而HERG突变体蛋白大部分滞留胞质。免疫印迹显示:不同于野生型HERG135 ku及155 ku 2个条带,突变体仅60 ku 1个条带。全细胞膜片钳检测WT+MT组IHERG较WT组下降55.23%(22.03±2.62 vs. 49.20±2.31 pApF, P<0.01)。结论HEK293细胞中杂合状态下截断突变体L539fs/47对野生型HERG电流的不完全负显性抑制作用。
关键词:HERG;无义突变;不完全负显性抑制作用;HEK293细胞
遗传性长QT综合征(long QT syndrome, LQTS)是一种由于编码心肌离子通道蛋白的基因突变所导致心肌细胞复极异常而引起的心脏离子通道病。LQTS易产生室性心律失常,尤其是尖端扭转型室性心动过速、晕厥和猝死[1]。最常见的LQT1~LQT3亚型中,我国以LQT2为主,占已知LQTS的45%。心室肌细胞复极最主要电流之一的快速激活延迟整流钾通道(rapidly activating delayed rectifier K+current,Ikr)[2],在调节心脏兴奋及维持心脏正常节律方面具有重要作用,其突变导致LQT2。人类ether- à-go-go相关基因(human-ether-á-go-go-related gene, HERG)编码1 159个氨基酸残基,组成Ikr通道的α亚基,经历两次糖基化后,4个α亚基组成四聚体,与KCNE2编码的起调节作用的β亚基共同组成Ikr;此外,HERG通道蛋白是几乎所有引起获得性LQTS药物结合的靶点[3]。
2007年,在一个LQTS家系中发现了父系遗传的大片段丢失的HERG基因无义突变体L539fs/47是已知第3例HERG大片段缺失[4]。HERG第7外显子cDNA第1 619~1 637核苷酸位点19个碱基缺失,同时1 692处有一单核苷酸多态性位点(single nucleotide polymorphism, SNP,A→G)。此突变及SNP导致第539位亮氨酸位点后第47个氨基酸提前出现终止密码子。HERG无义突变往往引起终止密码子提前出现,从而产生毒性的截短的通道蛋白,通过负显性机制导致LQT2的发生[5]。而另有研究,HERG蛋白C-端存在通道的组装识别信号,C-端突变或截短可引起组装识别信号丢失,不能组装成有功能的通道,可能出现单倍体不足[6]。HERG基因无义突变体L539fs/47的功能如何值得进一步研究,本文探讨无义突变体L539fs/47对野生型HERG电流的作用。
1材料与方法
1.1细胞野生型HERG稳转HEK293细胞(北京爱思益普公司李英骥博士提供),用DMEM高糖型培养基加100 mL/L胎牛血清在37 ℃,5 mL/L CO2条件下培养HEK293细胞。
1.2试剂与仪器质粒pcDNA3-HERG、pcDNA3-L539fs/47(本实验室张爱峰博士提供,已测序鉴定)、pRK5-GFP(美国Sigma公司)、HEK293细胞(本实验室提供)、DMEM培养基及胎牛血清(美国Hyclone公司)、X-treme GENE HP DNA转染试剂(美国Roche公司)、反转录试剂盒及实时荧光定量PCR(TaKaRa公司)、兔抗人HERG-N端单克隆抗体P0749(Sigma公司)、HRP标记的山羊抗兔IgG二抗(Thermo公司)、FITC标记的山羊抗兔IgG二抗(Bioworld公司)、Genshare超敏化学发光底物试剂盒(西安晶彩生物),其他均为国产分析纯试剂。2000型凝胶成像系统(美国Bio-Rad公司)、UV3000紫外可见分光光度仪(日本岛津公司)、PTC-200DNA扩增仪(美国MJ公司)、Bio-Rad/PAC/200W电泳仪(美国伯乐公司)、IMP-2型倒置相差生物显微镜(日本Olympus公司)、Axon-700单探头膜片钳放大器(美国Axon公司)、MX7600/R 右手显微操作系统(美国SD公司)、Model P-97程控微电极拉制仪(美国Sutter公司)、毛细玻璃管电极(美国Sutter公司)、pCLAMP9.2膜片钳数据处理软件(美国Axon公司)。
1.3方法
1.3.1细胞转染PCR及免疫荧光实验采用6孔板中HEK293细胞密度调整至40%~50%后,FuGENE®HD转染剂:质粒总量选取4 μL∶2 μg比例转染。膜片钳实验:一组稳转野生型HERG的HEK293细胞直接进行膜片钳试验;另一组6孔板中的稳转细胞瞬时转染pcDNA3-MT,转染预混物包括无血清DMEM 100 μL、转染剂4 μL,质粒分别2 μg(pcDNA3-MT∶示踪质粒pRK5-GFP=1.6 μg∶0.4 μg),转染36 h后进行膜片钳检测。
1.3.2实时定量PCRpcDNA3-L539fs/47(即pcDNA3-MT)0.75 μg或野生型pcDNA3-HERG(即pcDNA3-WT)、指示质粒pRK5-GFP 0.25 μg、无血清DMEM 50 μL、转染剂1 μL室温预混孵育15 min,加入HEK293细胞的24孔板中。转染36 h后,PBS冲洗3次。氯仿法提取RNA后37 ℃ 15 min,85 ℃ 5 min,4 ℃,逆转录为cDNA,引物:5′-ATCGGCAACATGGAGCAG-3′,5′-TGAGTTGGTGTTGGGAG-
AGAC-3′;内参GAPDH引物5′-TCATGGGTGTGAACCATGAGAA-3′;5′-GGCATGGACTGTGGTCATGAG-3′。扩增条件:95 ℃ 3 min;95 ℃ 10 s,55.9 ℃ 30 s,72 ℃ 30 s,共循环40次;72 ℃ 5 min。IQ5软件获取Ct值,通过相对表达量2-△△Ct进行统计。
1.3.3免疫荧光转染前培养基24孔板爬片上HEK293细胞密度调整至40%~50%。将pcDNA3-WT 0.75 μg、pcDNA3-MT分别与无血清DMEM 50 μL、转染剂1 μL室温预混孵育15 min,加入6孔板中。共转染36 h后免疫荧光染色。一抗(1∶25,PBS稀释)4 ℃,孵育14~18 h,FITC荧光标记二抗(1∶200,PBS稀释)避光37 ℃,孵育90 min后碳酸甘油(pH=9.2~9.4)缓冲液封片,在荧光显微镜下观察,波长488 nm蓝光激发出绿色荧光。
1.3.4免疫印迹野生组:将pcDNA3-WT转染进HEK293细胞,杂合组:将pcDNA3-WT联合pcDNA3-MT转染进HEK293细胞;36 h后提取蛋白变性后保存。浓缩胶80 V,20~30 min,80 g/L分离胶120 V,60~90 min,兔抗人一抗(1∶200稀释),4 ℃,过夜,TBST稀释HRP标记的二抗(山羊抗兔二抗1∶3 000稀释),室温2 h,HRP化学发光法显影。
1.3.5全细胞膜片钳检测采用稳定表达HERG-WT的HEK293细胞株,瞬时转染pcDNA3-L539fs/47。通过固定pcDNA3-HERG的表达,增加钳制细胞数,减少因瞬时转染pcDNA3- L539fs/47造成的误差。电极外液(NaCl 137 mmol/L、KCl 4 mmol/L、CaCl21.8 mmol/L、MgCl21 mmol/L、葡萄糖10 mmol/L、HEPES 10 mmol/L,NaOH调节pH=7.4),电极内液(KCl 130 mmol/L、MgCl21 mmol/L、EGTA 5 mmol/L、MgATP 5 mmol/L、HEPES 10 mmol/L,KOH调节pH=7.2)。夹取处理好的细胞爬片于培养皿中,细胞面向上;将充灌好电极内液的玻璃微电极移至细胞池内液面以下,阻抗为2~5 MΩ;选取膜折光性好,GFP荧光强度相近的细胞,电位钳制在-80 mV,与细胞封接阻抗达2~3 GΩ以上后,稳定30 s,快速负压破膜形成全细胞记录模式,刺激程序参考HUO等[7]。HERG通道电流由时间依赖性电流和尾电流两部分组成,本实验HERG电流均采用尾电流拟合。由Clampex 9.2软件及Axon700B放大器完成,信号经Axon700B放大器并经过A/D转换后(Digidata 1200A, Axon Instrument, USA)储存于计算机中。
2结果
2.1野生型与突变型HERG mRNA表达的比较以WT组的4.3 mmol/L亚组为对照组,计算出HERG的mRNA相对表达结果。基因型对HERG表达存在影响:WT组HERG表达量显著高于MT组(1.066±0.612vs. 0.254±0.397,P<0.01。图1)。

图1ERG-WT和突变体L539fs/47的RT-PCR结果
Fig.1 The RT-PCR of HERG-WT and mutant L539fs/47
A:溶解曲线和扩增曲线;B:PCR产物琼脂糖凝胶电泳;1、2、3:内参GAPDH PCR产物,146 bp;4、5、6:HERG PCR产物,198 bp;7:Marker。
2.2HEK293细胞中野生型HERG蛋白多于突变通道蛋白免疫荧光检测发现野生型HERG蛋白多于突变体,野生型HERG蛋白主要分布在细胞膜上;而HERG突变体蛋白大部分滞留在胞质(图2)。
2.3突变型HERG仅表达1条蛋白条带免疫印迹显示:不同于野生型HERG 135 ku及155 ku 2个条带,突变体仅60 ku 1个条带。共转染的WT+MT的杂合组出现135 ku、155 ku和60 ku 3个条带。WT组155 ku条带高于MT组中60 ku条带;杂合子中,155 ku成熟的野生型HERG表达明显多于60 ku的突变型HERG条带(图3)。
2.4细胞膜片钳检测结果程控刺激诱发尾电流可见野生组尾电流最大,突变组未诱发出明显的尾电流,提示突变型是无功能通道型,杂合组尾电流介于两者之间,提示通道功能受到抑制(图4)。在研究HERG的电生理功能时,选用尾电流,排除失活门引起的内向整流的干扰,其变化反应了通道的电压依赖性激活时程。MT组基本诱发不出尾电流IHERG。WT+MT组(n=20)IHERG较WT组(n=16)下降55.23%(22.03±2.62vs. 49.20±2.32 pApF,P<0.01,图5)。
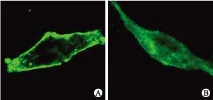
图2免疫荧光法检测HERG-WT及HERG-MT的表达
Fig.2 The immunofluorescence of HERG-WT and mutant L539fs/47
A:野生型HERG;B:突变体L539fs/47。
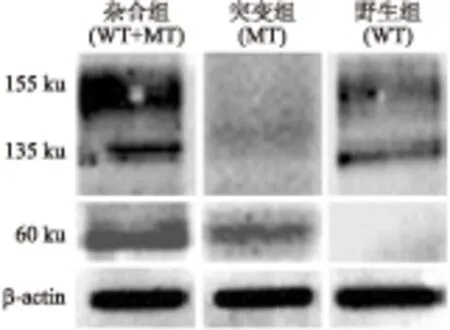
图3WT组和WT+MT组HERG蛋白的表达(免疫印迹)
Fig.3 The expression of HERG protein in WT group and WT+MT group (Western blot)
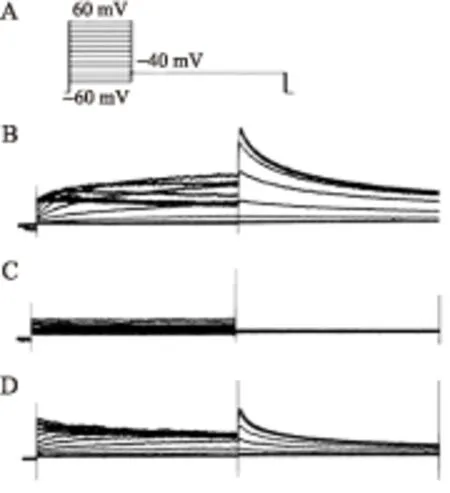
图4WT、MT和WT+MT组IHERG的尾电流
Fig.4 The tail currents ofIHERGin WT, MT and WT+MT groupsA:程控刺激图;B:野生组尾电流;C:突变组尾电流;D:杂合组尾电流。
3讨论
HERG蛋白的C-端的500个氨基酸与通道蛋白的稳定、组装和成熟相关。C-端丢失氨基酸超过311个,HERG通道功能丧失[9]。与此一致,L539fs/47的C-端完全丢失后,导致通道功能丧失,纯突变体通道无激活电流。
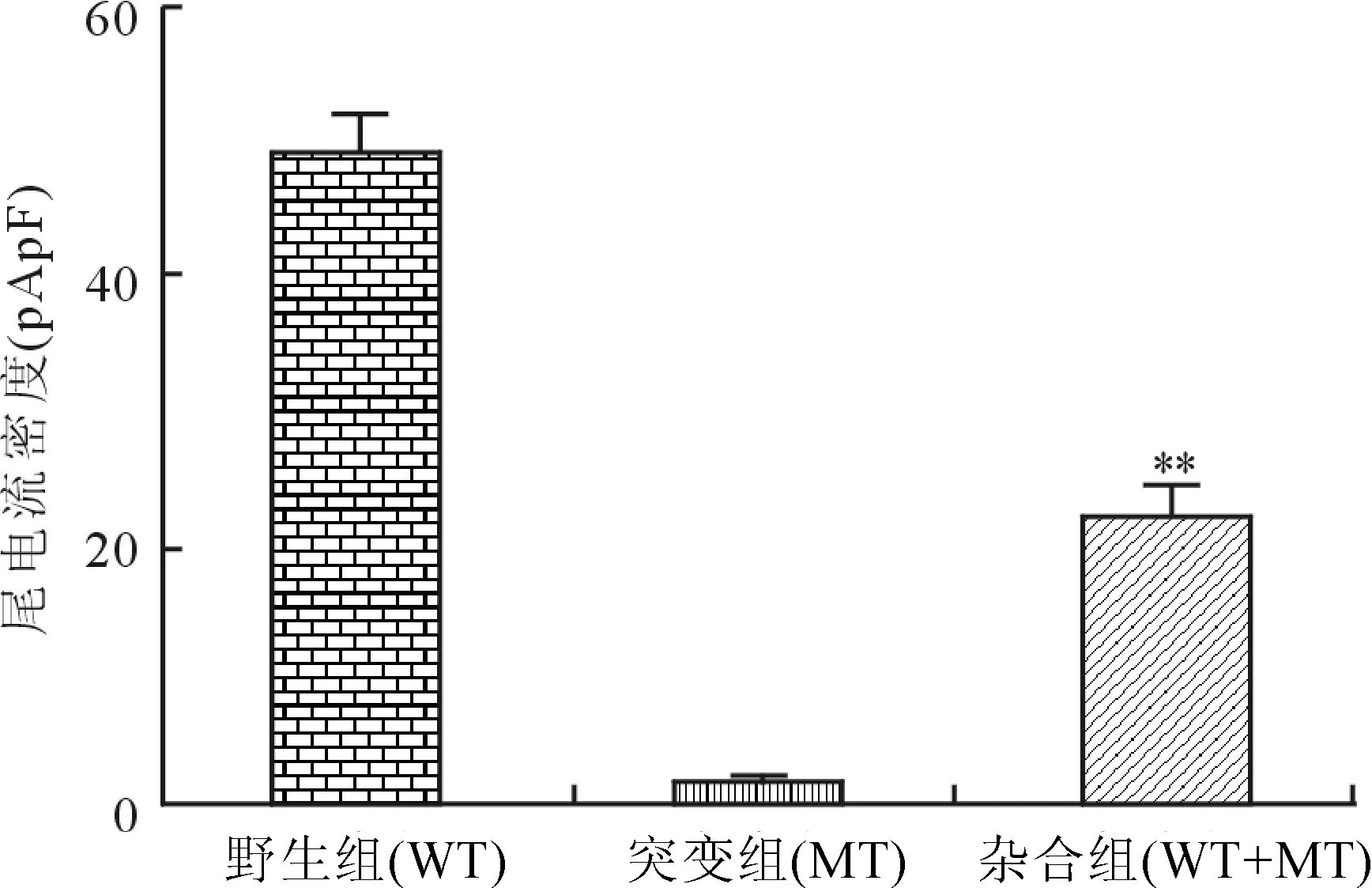
图5WT、MT和WT+MT组不同基因型间尾电流密度的比较
Fig.5 The densities of IHERGin WT, MT and WT+MT groups
与MT组比较,**P<0.01。
通常HERG突变导致的转运障碍,如能与野生型通道蛋白组装,不影响四聚体形成,减少通道功能大于50%,对WT通道产生负显性抑制作用[10];例如r1014x[11]。如截断蛋白无法与野生型通道蛋白组装成异源四聚体;仅野生型蛋白组装的同源四聚体至细胞膜发挥功能,促使通道功能丧失50%[12],导致单倍体功能不足,如q725x[13]。本研究中免疫荧光证实L539fs/47蛋白可部分转运至细胞膜,突变型可与野生型蛋白形成异源性四聚体,膜片钳实验显示模拟杂合子,突变体减少通道功能55.23%,表现为负显性抑制。与ZHANG等[14]前期研究接近。理论上起负显性抑制的突变体,仅存在于野生型同源四聚体,通道功能抑制率应达到或接近75%。而本研究中,L539fs/47突变通道蛋白表现的负显性抑制为不完全显性负抑制,介于负显性抑制与单倍体不足之间。
同大多数LQT2一样,L539fs/47无电流,存在部分转运障碍,产生不完全显性负抑制,可能与HERG-WT形成异源性四聚体及无义介导的mRNA降解(NMD)机制使截短蛋白表达减少,导致有功能的纯HERG-WT四聚体相对增多有关。细胞在进化过程中,NMD机制可以消除含有提前终止密码子的mRNA,减少毒性蛋白的表达。2007年GONG等[15]证实,HERG的过早截断突变体R1014X和W1001X发生无义介导的降解,LQT2的无义突变通过NMD机制引起mRNA降解,而不是生产截断蛋白。与此相符合,本研究中HERG无义突变体L539fs/47的mRNA量不到HERG-WT的1/3,大部分的HERG的mRNA可能通过NMD机制降解。免疫印迹实验中代表HERG-MT的60 ku条带灰度值明显低于代表HERG-WT的155 ku,支持L539fs/47可能通过NMD机制导致mRNA水平下降,进而影响突变体蛋白表达。
L539fs/47属于C端无义突变,对于LQT2家系的研究证实位于C末端区域携带移码/无义突变的LQT2患者通过NMD机制可能只引起比较温和的表型异常。本突变家系的先证者20岁后病情才发作,且其兄弟仅为致病基因的携带者但未出现临床症状,二者均为杂合子,可能与NMD机制有关。NMD可能是生物体在进化过程中形成的防止致命性严重后果的一种自我保护机制,也可能是一种更普遍的离子通道疾病的致病机制[16]。
此外,因丢失糖基化位点,不完全糖基化的L539fs/47蛋白易降解可能也是不完全显性抑制作用的原因之一。HERG蛋白在内质网上进行两次重要的糖基化过程,分别位于细胞外S5、S6连接区域,其中核心糖基化在N598区域完成,两次糖基化后完全成熟的HERG蛋白组成四聚体转运至细胞膜发挥生理功能。本研究通过免疫印迹发现L539fs/47蛋白仅60 ku一个条带,只经过一次糖基化,免疫荧光同时证明不完全糖基化的L539fs/47无义突变可部分定位于细胞膜上。这可能与THOMAS报道的核心糖基化对HERG蛋白的装配和转运并不具有决定性的作用有关[17]。尽管糖基化对功能HERG通道在细胞表面表达不是必须的,但非糖基化的HERG通道的周转率更快,N端糖基化对HERG通道稳定起着重要的作用[18]。L539fs/47截短蛋白丢失了N-糖基化的位点,也可能导致截短的通道蛋白不稳定,易降解,使得稳定的野生型同源四聚体蛋白在胞膜分布相对更多。
大多数异常分泌蛋白质滞留在内质网(ER),还可以引起ER相关的降解,如果突变影响到亚基间相互作用,无法屏蔽滞留信号RXR受体,导致单体滞留在ER,HERG-WT与突变亚基不能构成四聚体[19],导致单倍体不足。而损害ER输出信号,导致四聚体滞留在ER,在这种情况下会导致负显性表型。通常认为,HERG蛋白C-端存在通道的组装识别信号,C-端突变或截短可引起组装识别信号丢失,不能组装成有功能的通道。然而,近来研究认为存在C-端突变的HERG可以装配出四聚体。WT亚基与MT亚基形成异质性四聚体,然后在四聚体中的一些可能会输出ER,但速度较慢,导致部分显性抑制野生型通道的活动。结合L539fs/47呈不完全负显性抑制及本研究中免疫荧光示截短蛋白部分分布至细胞膜,WT亚基与L539fs/47亚基可能形成异质性四聚体,出现了有功能的ER输出信号,导致部分转运至细胞膜。同时,异源四聚体可能遮蔽滞留信号[20],允许ER输出信号引导四聚体前向转运。
综上所述,HERG突变体L539fs/47与野生型通道蛋白组装,不影响四聚体形成,减少通道功能大于50%,对WT通道产生负显性抑制作用。因无义介导的mRNA降解、异常蛋白滞留在内质网引起的相关降解、不完全糖基化引起的通道蛋白不稳定导致HEK293细胞中杂合状态下无义突变体L539fs/47对野生型HERG电流最终起到不完全负显性抑制作用。
参考文献:
[1] SMITH PL, BAUKROWITZ T, YELLEN G, et al. The inward rectification mechanism of the HERG cardiac potassium channel[J]. Nature, 1996, 379(6568):833-836.
[2] SANGUINETTI MC, TRISTANI-FIROUZI M. hERG potassium channels and cardiac arrhythmia[J]. Nature, 2006, 440(7083):463-469.
[3] GUO J,WANG TZ,LI X,et al. Cell surface expression of human ether-a-go-go-related gene (hERG) channels is regulated by caveolin-3 protein via the ubiquitin ligase Nedd4-2[J]. J Biol Chem, 2012, 287(40):33132-33141.
[4] 廉姜芳, 周建庆, 黄晓燕, 等. 长QT综合征KCNH2基因S4区新移码突变L539fs/47的研究[J]. 中华医学遗传学杂志, 2010, 27 (1): 77-80.
[5] STUMP MR, GONG Q, PACKER JD, et al. Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation[J]. J Mol Cell Cardiol, 2012, 53(5):725-33.
[6] SUN T, GUO J, SHALLOW H, et al. The role of monoubiquitination in endocytic degradation of human ether-a-go-go-related gene (hERG) channels under low K+conditions[J]. J Biol Chem, 2011, 286(8):6751-6759.
[7] HUO JH, ZHANG YM, HUANG N, et al. The G604S-hERG mutation alters the biophysical properties and exerts a dominant-negative effect on expression of hERG channels in HEK293 cells[J]. Pflugers Arch, 2008, 456(5):917-928.
[8] CHENG WH, WANG WY, ZHANG J, et al. State-dependent blockade of human ether-a-go-go-related gene (hERG) K(+) channels by changrolin in stably transfected HEK293 cells[J]. Acta Pharmacol Sin, 2010, 31(8):915-922.
[9] AYDAR E, PALMER C. Functional characterization of the C-terminus of the human ether-a-go-go-related gene K+channel (HERG)[J]. J Physiol, 2001, 534(1):1-14.
[10] STUMP MR, GONG Q, PACKER JD, et al. Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation[J]. J Mol Cell Cardiol, 2012, 53(5):725-733.
[11] GONG QM, ROBINSON JC, ZHOU Z, et al. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome[J]. Mol Cell Cardiol, 2004, 37(6):1225-1233.
[12] FURUTANI M, RUDEAU MC, HAGIWARA N, et al. Novel mechanism associated with an inherited cardiac arrhythmia--Defective protein trafficking by the mutant HERG (G601S) potassium channel[J]. Circulation, 1999, 99(17):2290-2294.
[13] GONG QM, VINCENT GM, HORNE BD, et al. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome[J]. Circulation, 2007, 116(1):17-24.
[14] ZHANG AF, ZHANG L, LV Y, et al. aG-X. L539 fs/47, a truncated mutation of human ether-a-go-go-related gene (hERG), decreases hERG ion channel currents in HEK 293 cells[J]. Clin Exp Pharmacol Physiol, 2013, 40(1): 28-36.
[15] ZHANG YM, WANG JL, CHANG SE, et al. The SCN5A mutation A1180V is associated with electrocardiographic features of LQT3[J]. Pediatr Cardiol, 2014, 35(2):295-300.
[16] GONG Q, STUMP MR, ZHOU Z, et al. Position of premature termination codons determines susceptibility of hERG mutations to nonsense-mediated mRNA decay in long QT syndrome[J]. Gene, 2014, 539(2):190-197.
[17] THOMAS D, KIEHN J, KATUS HA, et al. Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing[J]. Cardiovasc Res, 2003, 60(2):235-241.
[18] XIAO J, LUO X, LIN H, et al. MicroRNA miR-133 represses HERG K+channel expression contributing to QT prolongation in diabetic hearts[J]. J Biol Chem, 2007, 282(17):12363-12367.
[19] AYON RJ, FERNANDEZ RA, YUAN JX, et al. Mutant hERG channel traffic jam. Focus onPharmacological correction of long QT-linked mutations in KCNH2 (hERG) increases the trafficking of Kv11.1 channels stored in the transitional endoplasmic reticulum[J]. Am J Physiol Cell Physiol, 2013, 305(9):C916-918.
[20] ZERANGUE N, SCHWAPPACH B, JAN YN, et al. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K-ATP channels[J]. Neuron, 1999, 22(3):537-548.
(编辑韩维栋)
The mutant L539fs/47 has the effect of an incomplete dominant negative inhibition on wild type HERG current in HEK293 cells
LÜ Ying1, ZHANG Jun-bo2, ZHANG Ai-feng3, LIU Zhong-wei1, WANG Jun-kui1,HAN Wen-qi2, PAN Jun-qiang1, LI Guo-liang2, SUN Chao-feng2
(1. Department of Cardiology, Shaanxi Provincial People’s Hospital, Xi’an 710068;2. Department of Cardiology, the First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061; 3. Department of Cardiology, the Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710004, China)
ABSTRACT:ObjectiveTo study the effect of nonsense mutation of L539fs/47, when coexpressed with wild type human ether-a-go-go-related gene (HERG) current in HEK293 cells in the heterozygous state. MethodsThirty-six hours after transfection by liposome, wild-type HERG and its mutant HERG-L539fs/47 were transfected into HEK293 cells. HERG mRNA expressions were detected by real-time PCR; its protein localization and quantitative expression were detected by immunofluorescence and Western blotting, respectively. Current densities of HERG were detected by whole cell patch clamp. ResultsThe expression of HERG mRNA in wild-type group was higher than that in L539fs/47 mutation group (1.066±0.612 vs. 0.254±0.397, P<0.01). Immunofluorescence results showed that HERG protein was more than that in mutant group. The wild-type HERG protein was mainly distributed in the cell membrane; but retention of most mutant HERG protein L539fs/47 cytoplasm was detected. Western blotting showed that, different from the wild type HERG 135ku- and 155ku-bands, only one 60ku- band was detected in mutant group. Through the whole cell patch clamp test, the current density of HERG in heterozygous group (WT+MT group) decreased by 55.23% compared with WT group (22.03±2.62 vs. 49.20±2.31 pApF, P<0.01). ConclusionIn the heterozygous state, the truncation mutant L539fs/47 has the effect of incomplete dominant negative inhibition on wild type HERG current in HEK293 cells.
KEY WORDS:human ether-a-go-go-related gene (HERG); nonsense mutation; incomplete dominant negative inhibition; HEK293 cell
收稿日期:2015-05-22修回日期:2015-09-22
基金项目:国家自然科学基金资助项目(No.30800473)
通讯作者:孙超峰. E-mail: bobzjb@163.com.
中图分类号:R541.7
文献标志码:A
DOI:10.7652/jdyxb201603011
Supported by the National Natural Science Foundation of China (No.30800473)
优先出版:http://www.cnki.net/kcms/detail/61.1399.R.20160407.1712.008.html(2016-04-07)
