动态轴向压缩色谱制备高纯度断氧化马钱子苷的研究*
2016-06-05宋亚玲倪付勇王雪晶黄文哲王振中
宋亚玲,倪付勇,王雪晶,黄文哲,王振中,萧 伟**
(1.江苏康缘药业股份有限公司 连云港 222000;2.中药制药过程新技术国家重点实验室连云港 222000;3. 中药提取精制新技术重点研究室 连云港 222000)
动态轴向压缩色谱制备高纯度断氧化马钱子苷的研究*
宋亚玲1,2,3,倪付勇1,2,3,王雪晶1,2,3,黄文哲1,2,3,王振中1,2,3,萧 伟1,2,3**
(1.江苏康缘药业股份有限公司 连云港 222000;2.中药制药过程新技术国家重点实验室连云港 222000;3. 中药提取精制新技术重点研究室 连云港 222000)
目的:本研究旨在建立动态轴向压缩色谱法分离制备高纯度断氧化马钱子苷的方法。方法:金银花70%乙醇提取液先经大孔吸附树脂除杂富集后,采用动态轴向压缩色谱柱分离,以正相硅胶为填料,二氯甲烷-甲醇系统为洗脱剂,对断氧化马钱子苷进行精制纯化,分别运用HPLC、1H-NMR、13C-NMR和HR-ESI-MS对断氧化马钱子苷进行含量测定和结构表征。结果:产品经分析确定为断氧化马钱子苷,质量分数达98.76%。结论:采用动态轴向压缩工业色谱法制备断氧化马钱子苷的方法操作简便、快速、高效,为制备高纯度的断氧化马钱子苷提供了一条新途径。
断氧化马钱子苷 大孔树脂 动态轴向 工业色谱 高效液相色谱
断氧化马钱子苷(secoxyloganin)是环烯醚萜类化合物,是中药金银花Flos L. japonicae中的一个主要活性成分,该类成分具有抗病毒[1]、抗炎[2],抗菌[3]、抗血栓[4]、保肝利胆、增强免疫等作用,也可用于治疗心血管疾病、糖尿病及其并发症的原料药,具有很高的开发价值。金银花的常规鉴定是以其醇提物中的绿原酸为指标成分[5-8],但在其传统水煎剂中,发现含有大量环烯醚萜类化合物[9],因此绿原酸并不能代表金银花所含有的全部成分;故增加断氧化马钱子苷为质控指标,可以更全面地控制金银花药材质量。现有文献[9-11]中,关于断氧化马钱子苷的分离制备方法主要是萃取或树脂纯化以及硅胶、凝胶等反复纯化,步骤繁琐。因此,有必要寻找一种简便快速的制备方法,以便更好地开发利用断氧化马钱子苷。
本实验采用动态轴向压缩工业色谱技术,以正相硅胶为填料,实现断氧化马钱子苷的精制纯化,建立了一种快速大量制备高纯度断氧化马钱子苷的方法,且分离制备的断氧化马钱子苷达到了对照品的要求,为常规的分析检测和金银花药材及制剂的质量标准研究提供对照品,也为从其他植物中制备断氧化马钱子苷提供一条新的思路和方法。
1 材料与仪器
金银花药材经南京中医药大学吴启南教授鉴定为忍冬科植物忍冬L. japonicaThunb.的干燥花;断氧化马钱子苷为本实验室自制,纯度98.0%(江苏康缘药业股份有限公司,批号:131228)。D-101、HPD-100型大孔吸附树脂(天津海光化工有限公司);AB-8大孔树脂(南开大学化工厂);柱层析硅胶,200-300目(青岛海洋化工厂);甲醇,色谱纯(瑞典Oceanpak公司);甲酸,分析纯(南京化学试剂有限公司);95%乙醇,食用级(连云港长和酒业有限公司);双蒸水(自制)。Bruker-AV-400型核磁共振光谱仪(德国布鲁克公司);Agilent 1100高效液相色谱仪(美国Agilent公司),配自动进样器、四元泵、VWD检测器;AE240电子分析天平(瑞士Mettler公司);Agilent 1290-6538液质联用仪(美国Agilent公司);DAC-HB 80型动态轴向压缩工业色谱系统,80 mm×600 mm(江苏汉邦科技有限公司)。
2 方法与结果
2.1 色谱条件
色谱柱为Acuity C18柱(250 mm×4.6 mm,5 μm),流动相为甲醇(A)-0.1%甲酸溶液(B)。梯度洗脱程序如下:0-40 min,15%-50%A;40-45 min,50%-100%A。流速1.0 mL·min-1,进样体积10 μL,检测波长242 nm。
2.2 质谱条件
干燥气温度350℃,毛细管电压4 000 V,分离器电压65 V,传输电压135 V,质荷比扫描范围50-1 300,负离子模式,流动相甲醇-水(1:1),流速0.5 mL·min-1,不经过色谱柱。
2.3 提取工艺
取金银花10.0 kg,依次用10倍量、8倍量70%乙醇回流提取2次,合并提取液,滤过,滤液减压浓缩至相对密度1.05 g·mL-1(25℃)。搅拌加入乙醇至乙醇体积分数为80%,静置过夜,滤过,滤液减压浓缩至无醇味,加水稀释至1:1,静置过夜,滤过,弃去不溶物,即得金银花提取液,备用。断氧化马钱子苷的转移率为91.2%。
2.4 纯化工艺
2.4.1 树脂筛选
由于“2.3”项下制备的提取液含大量极性杂质成分,需要用树脂工艺纯化。考察D101、AB-8、HPD100 3种大孔树脂对样品中断氧化马钱子苷的吸附、解吸附效果。取3种经预处理的树脂各5 g(D101、AB-8、HPD100)置于100 mL具塞锥形瓶中,取金银花提取浓缩液加入瓶中,于室温静置24 h,吸附平衡后,滤去树脂,定量移取清液,测定断氧化马钱子苷的含量,计算吸附率。结果吸附率依次为72.1%、54.3%、62.7%,因此选择D101型大孔吸附树脂为应用树脂。
2.4.2 洗脱溶剂的筛选
取D101大孔吸附树脂40 g,取“2.3”项下的提取液100 mL上样,平行5份进行样品吸附后,水洗脱至Molish反应呈阴性后,分别用10%、20%、 30%、40%、60%乙醇溶液200 mL进行洗脱,收集洗脱液,减压浓缩并干燥,HPLC法测定,计算洗脱率及断氧化马钱子苷含量,结果见表1。
由表1可知,5倍树脂量10%乙醇洗脱液的断氧化马钱子苷含量最高,但洗脱不完全,30%、40%和60%乙醇洗脱液洗脱率虽高,但杂质也多,综合成本等因素考虑,确定选择20%乙醇作为洗脱溶剂,洗脱体积与树脂质量比为5:1。
2.4.3 树脂柱分离
2.4.3.1 树脂预处理
D101型大孔树脂用5%氢氧化钠溶液4 BV浸泡24 h,加4 BV水洗至中性,加5%盐酸溶液4 BV浸泡6 h,水洗至中性,加95%乙醇浸泡12 h后洗脱,至流出乙醇液与水混合不产生白色浑浊为止,用足量水洗至无醇味,备用。
2.4.3.2 吸附洗脱[12]
取预处理好的D101型大孔树脂柱(200 mm× 1 000 mm),取“2.3”项下提取液上样,吸附完成后先用水洗脱至Molish反应呈阴性后,再以5倍树脂量20%乙醇洗脱,收集20%乙醇洗脱液,减压浓缩,真空干燥,得断氧化马钱子苷粗品145.2 g(断氧化马钱子苷的质量分数为23.7%),备用。
2.5 动态轴向压缩色谱分离
2.5.1 柱填料的选择
实验以正相硅胶(200-300目)和十八烷基硅烷键合硅胶(YMG-C18,75 μm)为填料,进行色谱分离,结果表明正相硅胶不仅成本低而且有较好的分离度,得到的目标产品性状好,而十八烷基硅烷键合硅胶本身填料昂贵,并且所用色谱试剂成本太高,综合考虑选择以正相硅胶为分离填料。
2.5.2 色谱洗脱剂的选择[13]
以薄层色谱法(TLC)为依据,根据不同溶剂的性质,选择洗脱体系,比较分别以氯仿、二氯甲烷、乙酸乙酯与甲醇适当配比,对“2.4.3”项下的断氧化马钱子苷粗品进行单向展开,Rf值控制在0.2-0.8。结果表明,氯仿-甲醇、二氯甲烷-甲醇系统对样品均有较好的分离作用,结合工业化生产实际,最终选择二氯甲烷-甲醇系统作为色谱分离的洗脱剂。通过TLC最终确定柱色谱洗脱剂为二氯甲烷-甲醇系统,先用二氯甲烷-甲醇(12:1)洗脱除杂,然后用二氯甲烷-甲醇(9:1)洗脱,断氧化马钱子苷主要集中在9:1洗脱部位。
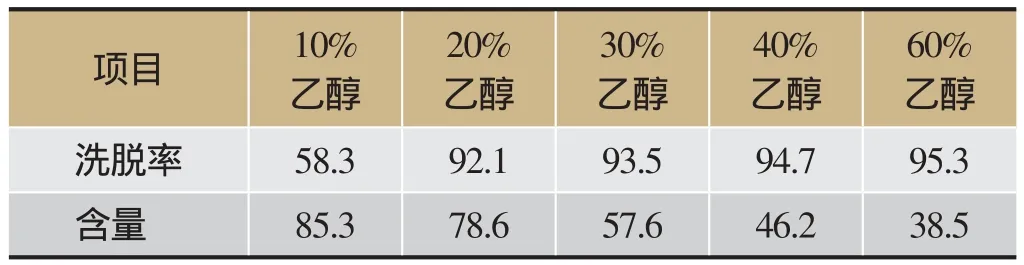
表1 不同浓度乙醇对断氧化马钱子苷的洗脱率和含量/%
2.5.3 正相工业色谱分离
采用动态轴向压缩工业色谱柱(80 mm×600 mm),以正相硅胶为填料制备纯化断氧化马钱子苷。将“2.4.3”项下断氧化马钱子苷粗品用流动相溶解,加适量硅胶拌样,干燥,干法上样,流动相为二氯甲烷-甲醇系统。洗脱程序如下:0-10 L,二氯甲烷-甲醇(12:1,V/V);10-20 L,二氯甲烷-甲醇(9:1,V/V)。体积流量30 mL·min-1,收集二氯甲烷-甲醇(9:1)洗脱的流分,浓缩干燥,即得白色粉末24.15 g,收率70.2%。
2.6 质量分数测定
2.6.1 对照品溶液的制备
精密称取断氧化马钱子苷对照品5.1 mg置于25 mL容量瓶中,用甲醇溶解定容至刻度,摇匀,即得。
2.6.2 供试品溶液的制备
精密称取“2.5.3”项下白色粉末5.3 mg至25 mL容量瓶中,用甲醇溶解稀释至刻度,摇匀,10 000 r·min-1离心5 min,取上清液,即得。
2.6.3 样品中断氧化马钱子苷的测定
分别取断氧化马钱子苷对照品和供试品溶液10 μL,按“2.1”项下色谱方法测定,用面积归一化法计算断氧化马钱子苷质量分数为98.76%,保留时间30.5 min。色谱图见图1-4,峰1为断氧化马钱子苷。
2.7 结构鉴定
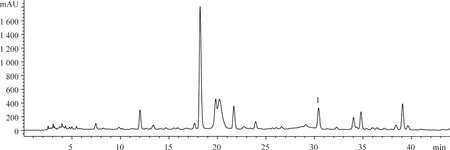
图1 金银花提取液的HPLC图

图2 D-101大孔树脂纯化后样品的HPLC图
断氧化马钱子苷,白色粉末(甲醇),HR-ESIMSm/z:403.126 [M-H]-,807.261 [2M-H]-(ESI-MS图见图5),分子式为C17H24O11。1H-NMR(400 MHz,CD3OD)δ:7.47 (1H,d,J=1.8 Hz,H-3),5.49(1H,d,J= 3.8 Hz,H-1),4.66(1H,d,J= 7.9 Hz,H-1′),3.68(3H,s,-OCH3),5.65(1H,ddd,J= 10.1,9.9,17.1 Hz,H-8),2.95(1H,dd,J= 16.5,4.9 Hz,H-6),2.27(1H,dd,J=9.0,16.5 Hz,H-6),2.82(1H,m,H-9).13C-NMR(100MHz,CD3OD)δ: 176.7(C-7),169.4(C-11),154.1(C-3),135.0(C-8),121.0(C-10),110.6(C-4),100.5(C-1′),98.1(C-1),78.9(C-5′),78.5(C-3′),75.1(C-2′),72.0(C-4′),63.2(C-6′),52.1(-OCH3),45.8(C-9),35.6(C-6),29.1(C-5)。以上数据与文献[14]报道一致,确定其为断氧化马钱子苷(secoxyloganin)。

图3 工业色谱纯化后样品的HPLC图

图4 断氧化马钱子苷对照品的HPLC图

图5 断氧化马钱子苷供试品溶液的ESI-MS图
3 讨论
由于断氧化马钱子苷具有较强的药理活性,对该化合物的研究具有较大的实用价值。考虑到大规模制备成本和分离效果两方面因素,本实验以金银花为原料,提取液先经醇沉、水沉及大孔树脂等工艺除去大量杂质,然后采用动态轴向压缩工业色谱分离制备断氧化马钱子苷,制备速度快、效率高,克服了传统硅胶柱色谱洗脱时间长、有机溶剂用量大、得率低等缺点,且制备量大;而且以正相硅胶为填料不仅成本低而且有较好的分离度,得到的断氧化马钱子苷纯度高。本方法操作简单,重现性好。
断氧化马钱子苷为金银花中的活性成分,但是在药材中含量并不是很高,因此该化合物的标准品比较昂贵,直接用来研究其药效、药理活性等成本较高,因此本研究建立了一种快速大量制备高纯度的断氧化马钱子苷的方法,分离制备的断氧化马钱子苷质量分数达98.76%,符合对照品的要求,为断氧化马钱子苷的制备提供了技术参考,对新药研究及进一步控制金银花药材及其制剂的质量标准研究具有重要意义。
1 Chen J L, Blanc P, Stoddart C A,et al. New Iridoids from the Medicinal Plant Barleriaprionitis with potent activity against respiratory syncytial virus.J Nat Prod, 1998, 61(10): 1295-1297.
2 李医明,曾华武,贺祥,等.玄参中环烯醚萜甙和苯丙素甙对LTB4产生及血小板聚集的影响.第二军医大学学报, 1999, 20(5): 301-303.
3 Tasdemir D, Scapozza L, Zerbe O,et al. Iridoid glycosides of leonuruspersicus.J Nat Prod, 1999, 62(6): 811-816.
4 Suzuki Y, Kondo K, Ikeda Y,et al. Antithrombotic effect of geniposide and genipin in the mouse thrombosis model.Planta Med, 2001, 67(9): 807-810.
5 国家药典委员会.中华人民共和国药典(一部).北京:化学工业出版社, 2010: 205-206.
6 邢俊波,李萍,刘云.不同产地、不同物候期金银花中绿原酸的动态变化研究.中国药学杂志, 2003, 38(1): 19-21.
7 高红宇,金万勤,郭立玮.不同精致工艺对金银花水提液中绿原酸含量的影响.世界科学技术-中医药现代化, 2010, 12(2): 241-244.
8 高晓艳,凌娅,萧伟. 不同产地加工方法的金银花中绿原酸、木犀草苷含量及指纹图谱比较. 世界科学技术-中医药现代化, 2010, 12(2): 291-293.
9 毕跃峰,田野,裴姗姗.金银花中裂环环烯醚萜苷类化学成分研究.中草药, 2008, 39(1): 18-21.
10 田野.金银花化学成分研究.郑州:郑州大学硕士学位论文, 2007.
11 陈雨,赵友谊,吴双,等.灰毡毛忍冬花蕾水溶性化学成分研究.中药材, 2012, 35(2): 231-234.
12 朱浩,侯世祥,孙毅毅,等.大孔吸附树脂吸附纯化不同中药有效部位特性研究.中国中药杂志, 1998, 23(10): 607-609.
13 王倩,朱靖博,顾丰颖,等.萃取与工业色谱相结合批量制备丹参中丹酚酸B.中国中药杂志, 2007, 32(21): 2317-2318.
14 李畅,戴毅,张金博,等.金银花中1个新的环烯醚萜苷类化合物.中草药, 2013, 44(21): 2951-2954.
Isolation of High-Purity Secoxyloganin from Flos Lonicera Japonica by Industrial Dynamic Axial Compression Preparative Chromatography
Song Yaling1,2,3, Ni Fuyong1,2,3, Wang Xuejing1,2,3, Huang Wenzhe1,2,3, Wang Zhenzhong1,2,3, Xiao Wei1,2,3
(1.Jiangsu Kanion PharmaceuticalCo., Ltd., Lianyungang 222000, China; 2.State Key Laboratory of New-tech for Chinese Medicine Pharmaceutical Process, Lianyungang 222000, China; 3. The Key Laboratory for the New Technique Research of TCM Extraction and Purification, Lianyungang 222000, China)
This study was aimed to establish an industrial chromatographic separation technology of high-purity secoxyloganin. The 70% alcohol extract of Flos L. japonica was separated and concentrated by macroporous resin and industrial chromatography. Normal phase silica gel was taken as solid phase, and methylene chloridemethanol system as eluent for the dynamic axial compression column. The determination and characterization of secoxyloganin was detected by HPLC,1H-NMR,13C-NMR and HR-ESI-MS. The results showed that the content of secoxyloganin reached to 98.76%. It was suggested that the industrial chromatographic separation technology of secoxyloganin was convenient, rapid and efficient, which provided a new method for the preparation of highpurity ecoxyloganin.
Secoxyloganin, macroporous resin, dynamic axial compression, industrial chromatography, HPLC
10.11842/wst.2016.03.031
R284
A
(责任编辑:朱黎婷 张志华,责任译审:朱黎婷)
2014-10-28
修回日期:2014-11-07
* 科学技术部“重大新药创制”科技重大专项(2013ZX09402203):现代中药创新集群与数字制药技术平台,负责人:王振中。
** 通讯作者:萧伟,本刊编委,研究员级高级工程师,博士,主要研究方向:中药新药的研究与开发。
