生物催化还原亚胺类化合物制备手性胺的研究进展
2016-05-31陈永正
陈永正
(遵义医学院 药学院,贵州 遵义 563099)
专家论坛
生物催化还原亚胺类化合物制备手性胺的研究进展
陈永正
(遵义医学院 药学院,贵州 遵义563099)
[摘要]近些年,亚胺的不对称催化还原制备手性胺逐渐成为有机合成中的一个研究热点,其方法主要包括金属催化、有机小分子催化和生物催化的不对称还原。本文着重介绍生物催化还原方法中亚胺还原酶的筛选、相关基因的克隆表达以及用于亚胺还原的人工金属酶的研究进展。
[关键词]生物催化;不对称还原;亚胺还原酶;人工金属酶
氨基广泛存在于自然界各种具有生物活性的天然产物中,如:生物碱(吗啡、麻黄碱)、蛋白质、激素以及抗生素等。其中,手性胺作为一类极其重要的化合物,不仅可以作为一些药物合成的中间体和农用化学品的有效成份,也可以作为一些过渡金属的配体或者直接作为有机小分子催化剂用于催化手性化合物的合成,同时还可以用于香料香精产业[1-2]。因此,对手性胺合成的研究受到许多化学家的高度关注,目前其合成方法主要包括化学催化合成法、生物酶催化合成法及组合催化合成法,尤其对化学催化潜手性C=N键的不对称还原反应的研究较为透彻,底物适用范围广泛,涵盖N-芳基亚胺、N-烷基亚胺、烯胺等。目前使用的化学催化剂包括手性有机小分子催化剂,如手性双烯-硼烷[3]、手性磷酸[4-6];金属催化剂:如铱[7-11]、钯[12]、钛[13]、铑[14-15]、铼[16]、钌[17]、钴[18]。
然而,随着制药工业对环境以及行业规范要求的日益提高,传统化学合成的方法局限性日益凸显,生物催化的不对称合成因具有反应条件温和、选择性高、绿色无污染等优点,并且随着分子生物学技术的发展,对酶的修饰和改性变得更加容易,逐渐被广泛应用于手性胺的合成中。例如Christopher K S.小组采用高活性的转氨酶突变体催化酮到手性胺的不对称转化,成功实现了西格列汀的合成[19];氰基还原酶作为一种新型的生物催化剂用于氰基化合物的不对称还原,可以为手性胺的合成提供了一种新方法[20-23]。此外,利用基因工程技术对依赖于NADPH辅酶的氨基酸脱氢酶[24]与红球菌属中的苯丙氨酸脱氢酶[25]进行改造,获得的突变体酶可以催化酮的不对称还原胺化反应,用于合成手性胺。
在生物体中也存在亚胺还原的生化反应过程,例如:在生物体内存在一种“一碳单位”的接纳体和供体-叶酸是一种水溶性B族维生素,广泛存在于绿色植物、蘑菇、酵母、肝脏和肾脏中。以NADPH为辅酶,叶酸 (1-a)能在二氢叶酸(DHF)还原酶作用下,经两次连续还原,先后生成7,8-二氢叶酸 (1-b)和5,6,7,8-四氢叶酸 (1-c),即辅酶F (见图1)。
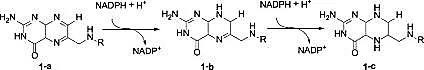
图1 二氢叶酸还原酶反应
此外,在生物体内氨基酸代谢的过程中,由谷氨酸脱氢酶 (GDH)介导的氨基酸的氮转移过程中可以产生亚胺中间体,然后经不对称还原得到手性α-氨基酸 (见图2)[26]。

图2 GDH介导的氨基酸代谢过程
尽管亚胺还原酶催化亚胺的不对称还原反应早有相关报道,但由于传统亚胺化合物在水相体系中差的稳定性,易分解生成相应的酮和胺,而水相往往又是生物酶催化反应的传统介质,这无疑限制了亚胺还原酶在催化亚胺不对称还原反应中的应用。大多数亚胺中间体为环状酮亚胺,并且与酮之间存在动态平衡,在酶催化作用下,平衡可以向生成新的手性氨的反应方向移动。因此,可以用醛或酮代替亚胺,在转氨酶催化作用下,实现手性胺的高对映选择性合成,从而克服以亚胺作底物在水溶液中不稳定的局限性。下面将重点对生物催化亚胺合成手性胺化合物的方法进行讨论。
1亚胺还原酶
2004年,Stephens G.小组以正丁醛、苯甲醛、正丁胺和苯胺作为底物,采用动态组合筛选的方法,以水和正十四烷作为两相反应体系,成功筛选出亚胺还原酶产生菌株Acetobacteriumwoodii。在筛选过程中他们发现,用咖啡酸酯诱导后的菌株在催化N-亚苄基苯胺和N-亚丁基苯胺的还原反应中,能够显示出高的催化活性;而不经诱导的菌株只能用于N-亚丁基苯胺和苯甲醛的还原,对其他底物的还原没有催化活性 (见图3)[27]。

图3 动态组合法在亚胺还原酶筛选中的应用
2008年,Chadha A.小组以芳香亚胺为底物,采用CandidaparapsilosisATCC 7330整细胞生物催化剂,实现了水相体系中亚胺的不对称还原反应,获得了R构型的芳香仲胺。研究过程中,他们发现苯环上连有吸电子基团或供电子基团对反应结果影响不大,对映选择性为95%~99%,产率为55%~80% (见图4)[28]。

图4 ATCC 7330催化芳香亚胺合成手性仲胺
2010年,Mitsukura K.小组以2-甲基-1-二氢吡咯 (2-MPN)为底物,从226株酵母菌、261株细菌、117株放线菌和84株真菌中筛选获得了五株具有亚胺还原活性的放线菌属微生物。用其中一株催化2-MPN的不对称还原,可以得到S构型的产物,而用另外四株催化该反应,则得到R构型的产物。值得注意的是,这些菌株具有较好的底物耐受性,即使在27.5~91 mmol/L的底物浓度下,产物的收率和ee值也可分别达到92%和99.2% (见图5)[29-30]。

图5 亚胺还原酶催化2-MPN的不对称还原
随后,他们克隆表达了菌株中的亚胺还原酶基因,发现对R/S选择性的亚胺还原酶均依赖辅酶NADPH,且均为同源二聚体结构,亚基大小分别为32 kDa、30.5 kDa。更有趣的是,在中性pH条件下,具有R选择性的还原酶对2-MPN表现出还原活性,在碱性条件下,则对 (R)-2-MP表现出氧化活性。此外,通过对两种不同选择性的亚胺还原酶进行氨基酸序列分析,发现S选择性的亚胺还原酶与6-磷酸葡萄糖酸脱氢酶具有60%的相似度,而R选择性的亚胺还原酶与6-磷酸葡萄糖酸脱氢酶的相似度只有37%[31]。
同年,Santos L S.小组采用Saccharomycesbayanus整细胞催化的方法,实现了β-咔啉亚胺类化合物的不对称还原,实验结果表明,R取代基对反应的选择性有着很大的影响。当R为C1-C11的脂肪族取代基时,该酶显示出S选择活性,当R取代基碳链长度增加到C15以上或者为芳香取代基团时,则酶显示出R选择活性[32]。在研究过程中,他们还发现蚯蚓细胞的提取液也可以用于β-咔啉亚胺的不对称还原反应。与之前不同的是,该生物催化剂只是显示出R选择性,产物ee值为85%~99% (见图6)[33]。
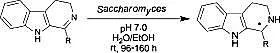
图6 整细胞催化β-咔啉亚胺类化合物不对称还原
2012年,Lamb A. L.小组从Yersiniaenterocolitica中分离得到依赖NADPH的亚胺还原酶,在噬铁耶尔森杆菌素形成过程中用于介导二氢噻唑的还原。通过将分离获得的亚胺还原酶与载脂蛋白及NADP+结合得到的复合物的结构进行测定,分析发现其结构与葡萄糖-果糖氧化还原酶、1,5-果糖苷还原酶以及胆绿素还原酶的结构类似。此外,他们还发现该酶的C端区域存在大量的特有的环状片段,这些特殊片段与来自Pseudomonasaeruginosa的噻唑啉亚胺还原酶的具有较高同源性,该结构域并不存在于其他结构类似物中,因此,他们认为该环状区域在底物识别过程中起着至关重要的作用。并且还对该结构作了进一步的解析,初步确定101号位的组氨酸和128号位酪氨酸以及NADPH分别在催化C=N双键还原的过程中起到提供质子的作用[34]。
2013年,Grogen G.小组用从Streptomyceskanamyceticus中获得的氧化还原酶(Q1EQE0),实现了单环亚胺2-甲基-1-二氢吡咯啉的不对称还原反应。Q1EQE0的分子量大小为32 kDa,在以NADPH作为辅酶的情况下,Km值达到8.21±1.07 mM,kcat值达到 (0.018±1.4)×10-3s-1,对映选择性大于99%。另外,当底物浓度达到30 mM以上时,反应表现出明显的底物抑制现象,经计算Ki为56.5±10.7 mM。当向反应体系中加入NADH时,则酶没有催化活性,此结果表明该酶对NADPH有着严格的依赖性。通过对其结构分析,他们发现Q1EQE0是以二聚体的形式催化反应的,其每个单体由28个氨基酸组成,其中187号天冬氨酸残基在酶的催化活性中起到关键性作用[35]。
2014年,Höhne M.小组分别从类芽孢杆菌PaenibacilluselgiiB69、链霉菌Streptomycesipomoeae91-03、假单胞菌PseudomonasputidaKT2440中克隆表达出三种亚胺还原酶,其中从假单胞菌中克隆获得的亚胺还原酶显示出最好的催化活性。进一步的基因突变证明该酶起催化作用的残基为组氨酸残基,有别于已报道的克隆于Streptomyceskanamyceticus的还原酶,后者起催化作用的是天冬氨酸残基 (见图7)[36]。
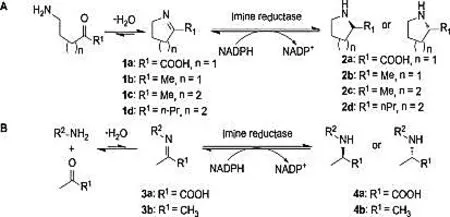
图7 还原酶催化合成光学纯的二次循环胺亚胺
同年,Hauer B.小组通过将已报道的亚胺还原酶基因的序列于已获得的亚胺还原酶序列进行比对分析,分别筛选出了具有R选择性和S选择性的亚胺还原酶的基因序列。通过进一步克隆表达、蛋白纯化获得了纯酶催化剂,并以此催化2-甲基-二氢吡咯的不对称还原反应。研究表明,与之前报道的亚胺还原酶相比,这些酶可以表现出更好的催化活性,而且活性中心的氨基酸残基对还原活性的维持起着至关重要的作用[37]。
2人工金属酶
除了上述的亚胺还原酶外,人工金属酶也被广泛应用于亚胺的不对称还原反应,这类催化剂主要是利用肽支架与化学催化剂的中心结合,以达到改变反应的微环境的目的,实现对反应选择性的控制。
2011年,Ward T. R.小组利用生物素衍生的双胺配体/金属配合物与野生型链霉亲和素蛋白协同催化6,7-二甲氧基-1-甲基-3,4-二氢异喹啉的不对称还原反应,得到了R构型的猪毛菜定[38]。后来,他们采用相同的技术得到了人造转移氢化酶,并将其用于生物催化的氧化还原级联反应。通过将此酶分别与NADH依赖、FAD依赖和血红素依赖的酶结合,以此考察酶的催化效率,用于证明该方法的普适性[39]。进一步将酶活性部位插入疏水性氨基酸残基,得到新的人工金属酶[40],其催化效率大大提高,较野生型亚胺还原酶的催化效率提高7倍,反应的对映选择性也得到了提高,并且还很好地克服了底物抑制的局限性。即使将底物浓度提高到150 mmol/L,反应的Kcat也能达到20 min-1。
在前期的工作基础上,他们通过基因工程技术对宿主蛋白周围的基因进一步修饰,筛选获得的氢化酶 (ATHase),能够高效催化6,7-二甲氧基-1-甲基-3,4-二氢异喹啉的不对称转移氢化反应,得到不同构型的还原产物。进一步研究证明,金属铱和链霉亲和素蛋白的结合比例可以在很大程度上影响亚胺还原酶的动力学参数及其催化反应的对映选择性。通过计算机辅助计算和X射线衍射技术分析有机金属与生物支架的对接情况,并结合饱和动力学研究,他们提出了高对映选择性“诱导锁钥”假说,即宿主蛋白的结构决定了金属铱辅因子的组态,该组态又在很大程度上决定了亚胺还原酶的选择性。更值得一提的是,人工金属酶被证明具有野生酶的原始活性,其不仅可以弥补野生酶的不足,还可用于催化串联反应 (见图8)[41-42]。
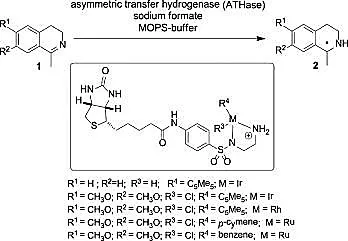
图8 人工亚胺还原酶催化的不对称转移氢化
3展望
本文对亚胺还原酶制备手性胺的应用研究进行了简要的概述,重点探讨了几个课题组将生物催化法用于亚胺的不对称还原制备光学纯仲胺的研究进展。综观此领域的研究不难发现,目前采用亚胺还原酶合成手性胺的报道仍然较少,其主要归结于亚胺还原酶的类型及其催化反应底物的普适性仍有较大的局限性。近来,对生物体中的代谢过程已经有了比较深入的研究,并且最新发现的几个氧化还原酶可以在非水介质中进行有效的生物催化反应[43],这一发现为实现非水介质中的生物催化亚胺还原合成手性胺提供了可行性参考。因此,如何更好的利用这些最新研究成果,深入开发丰富微生物资源中的亚胺还原酶,并进一步运用分子生物学和基因工程技术对亚胺还原酶进行改造,对于丰富亚胺还原酶的种类和构建一些新型的化学合成反应,实现更广泛的亚胺底物的不对称还原,仍然是一项具有一定挑战性且极有意义的工作,也将会受到越来越多的关注。
[参考文献]
[1] Breuer M, Ditrich K, Habicher T, et al. Industrial methods for the production of optically active intermediates [J]. Angew Chem Int Ed, 2004, 43(7): 788-824.
[2] Nugenta T C, El-Shazly M. Chiral amine synthesis: recent developments and trends for enamide reduction, reductive amination, and imine reduction [J]. Adv Synth Catal, 2010, 352(5): 753-819.
[3] Liu Y B, Du H F. Chiral dienes as “ligands” for borane-catalyzed metal-free asymmetric hydrogenation of imines [J]. J Am Chem Soc, 2013, 135(18): 6810-6813.
[4] Rueping M, Sugiono E, Azap C,et al.Enantioselective brønsted acid catalyzed transfer hydrogenation:organocatalytic reduction of imines [J]. Org Lett, 2005, 7(17): 3781-3783.
[5] Wen W, Zeng Y,Peng L Y,et al.Asymmetric synthesis of α-amino ketones by brønsted acid catalysis [J]. Org Lett,2015, 17(15): 3922-3925.
[6] Simón L, Goodman J M. Theoretical study of the mechanism of hantzsch ester hydrogenation of imines catalyzed by chiral BINOL-phosphoric acids [J]. J Am Chem Soc, 2008, 130(27): 8741-8747.
[7] Guo C, Sun D W, Yang S, et al. Iridium-catalyzed asymmetric hydrogenation of 2-pyridyl cyclic imines: A highly enantioselective approach to nicotine derivatives [J]. J Am Chem Soc, 2015, 137(1):90-93.
[8] Hou G H, Tao R, Sun Y K, et al. Iridium-monodentate phosphoramidite-catalyzed asymmetric hydrogenation of substituted benzophenone N-H imines [J]. J Am Chem Soc, 2010, 132(7): 2124-2125.
[9] Hou G H, Gosselin F, Li W. Enantioselective hydrogenation of N-H imines [J]. J Am Chem Soc, 2009, 131(29): 9882-9883.
[10] Li C Q, Wang C, Marcos B V, et al. Chiral counteranion-aided asymmetric hydrogenation of acyclic imines [J]. J Am Chem Soc,2008, 130(44): 14450-14451.
[11] Mršiĉ N, Minnaard A J, Feringa B L, et al. Iridium/monodentate phosphoramidite catalyzed asymmetric hydrogenation of N-aryl imines [J]. J Am Chem Soc, 2009, 131(24): 8358-8359.
[12] Chen M W, Duan Y, Chen Q A, et al. Enantioselective Pd-catalyzed hydrogenation of fluorinated imines: facile access to chiral fluorinated amines [J]. Org Lett, 2010, 12(21): 5075-5077.
[13] Hansen M C, Buchwald S L. A method for the asymmetric hydrosilylation of N-aryl imines [J]. Org Lett, 2000, 2(5): 713-715.
[14] Li C Q, Xiao J L. Asymmetric hydrogenation of cyclic imines with an ionic Cp*Rh(III) catalyst [J]. J Am Chem Soc, 2008, 130(40): 13208-13209.
[15] Shende V S, Deshpande S H, Shingote S K, et al. Asymmetric transfer hydrogenation of imines in water by varying the ratio of formic acid to triethylamine [J]. Org Lett, 2015, 17(12): 2878-2881.
[16] Nolin K A, Ahn R W, Toste F D. Enantioselective reduction of imines catalyzed by a rhenium(V)-oxo complex [J]. J Am Chem Soc, 2005, 127(36): 12462-12463.
[17] Rashid K A, Lough A J, Morris R H. RuHCl (diphosphine) (diamine):catalyst precursors for the stereoselective hydrogenation of ketones and imines [J]. Organometallics, 2001, 20(6): 1047-1049.
[18] Valencia M A, Cabrera A. The first example of asymmetric hydrogenation of imines with Co2(CO)8/(R)-BINAP as catalytic precursor [J]. J Mol Catal A: Chem, 2013, 366(1): 17-19.
[19] Savile C K, Janey J M, Mundorff E C, et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture [J]. Science, 2010, 329(5989): 305-309.
[20] Domínguez de M P. Nitrile Reductases: A forthcoming wave in biocatalysis [J]. Chem Cat Chem, 2011, 3(11): 1683-1685.
[21] Moeller K, Nguyen G S, Hollmann F, et al. Expression and characterization of the nitrile reductase queF fromE.coli[J]. Enzyme Microb Technol, 2013, 52(3): 129-133.
[22] Wilding B, Winkler M, Petschacher B, et al. Targeting the substrate binding site ofE.colinitrile reductase queF by modeling, substrate and enzyme engineering [J]. Chem Eur J, 2013, 19(22): 7007-7012.
[23] Wilding B, Winkler M, Petschacher B, et al. Nitrile reductase from geobacillus kaustophilus: A potential catalyst for a new nitrile biotransformation reaction [J]. Adv Synth Catal, 2012, 354(11-12): 2191-2198.
[24] Abrahamson M J, Vázquez F E, Woodall N B, et al. Development of an Amine Dehydrogenase for Synthesis of Chiral Amines [J]. Angew Chem Int Ed, 2012, 51(16): 3969-3972.
[25] Ye L J, Toh H H, Yang Y, et al. Engineering of amine dehydrogenase for asymmetric reductive amination of ketone by evolving Rhodococcus phenylalanine [J]. ACS catal, 2015, 5(2): 1119-1122.
[26] Hallen A, Jamie J F, Copper A J L. Imine reductases: a comparison of glutamate dehydrogenase to ketimine reductases in the brain [J]. Neurochem Res, 2014, 39(3): 527-541.
[27] Li H, Williams P, Micklefield J, et al. A dynamic combinatorial screen for novel imine reductase activity [J]. Tetrahedron, 2004, 60(3): 753-758.
[28] Vaijayanthi T, Chadha A, et al. Asymmetric reduction of aryl imines using Candida parapsilosis ATCC 7330 [J]. Tetrahedron: Asymmetry, 2008, 19(1): 93-96.
[29] Mitsukura K, Suzuki M, Nagasawa T, et al. Asymmetric synthesis of chiral cyclic amine from cyclic imine by bacterial whole-cell catalyst of enantioselective imine reductase [J]. Org Biomol Chem, 2010, 8(8): 4533-4536.
[30] Mitsukura K, Suzuki M, Nagasawa T, et al. Purification and characterization of a novel (R)-imine reductase fromStreptomycessp. GF3587 [J]. Biosci Biotechnol Biochem, 2011, 75(9): 1778-1782.
[31] Mitsukura K, Kuramoto T, Nagasawa T, et al. A NADPH-dependent (S)-imine reductase (SIR) fromStreptomycessp. GF3546 for asymmetric synthesis of optically active amines: purification, characterization, gene cloning, and expression [J]. Appl Microbiol Biotechnol, 2013, 97(18): 8079-8083.
[32] Espinoza M M, Petta T, Santos L S, et al. Bioreduction of β-carboline imines to amines employingSaccharomycesbayanus[J]. Tetrahedron: Asymmetry, 2010, 21(16): 1988-1992.
[33] Gallardo Y M, Soriano M P C, Santos L S. Stereoselective bioreduction of β-carboline imines through cell-free extracts from earthworms (Eiseniafoetida) [J]. Tetrahedron: Asymmetry, 2013, 24(8): 440-443.
[34] Keneely K M, Lamb A L. Two structures of a thiazolinyl imine reductase fromYersiniaenterocoliticaprovide insight into catalysis and binding to the nonribosomal peptide synthetase module of HMWP1 [J]. Biochem, 2012, 51(44): 9002-9013.
[35] Rodríguez M M, Frank A, Grogan G, et al. Structure and activity of NADPH-dependent reductase Q1EQE0 fromStreptomyceskanamyceticus, which catalyses theR-selective reduction of an imine substrate [J]. Chem Bio Chem, 2013, 14(11): 1372-1379.
[36] Gand M, Muller H, Höhne M, et al. Characterization of three novel enzymes with imine reductase activity [J]. J Mol Catal B: Enzym, 2014, 110(8): 126-141.
[37] Scheller P N, Fademrecht S, Hofelzer S, et al. Enzyme toolbox: novel enantio complementary imine reductases [J]. Chem Bio Chem, 2014, 15(15): 2201-2204.
[38] Dürrenberger M, Heinisch T, Wilson Y M, et al. Artificial transfer hydrogenases for the enantioselective reduction of cyclic imines [J]. Angew Chem Int Ed, 2011, 50(3): 3026-3029.
[39] Köhler V, Wilson Y M, Dürrenberger M, et al. Synthetic cascades are enabled by combining biocatalysts with artificial metalloenzymes [J]. Nat Chem, 2013, 5(2): 93-99.
[40] Schwizer F, Kohler V, Durrenberger M, et al. Genetic optimization of the catalytic efficiency of artificial imine reductases based on biotin-streptavidin technology [J]. ACS Catal, 2013, 3(8): 1752-1755.
[41] Robles V M, Durrenberger M, Heinisch T, et al. Structural, kinetic, and docking studies of artificial imine reductases based on biotin-streptavidin technology: An induced lock-and-key hypothesis [J]. J Am Chem Soc, 2014, 136(44): 15676-15683.
[42] Robles V M, Vidossich P, Lledoós A, et al. Computational insights on an artificial imine reductase based on the biotin-streptavidin technology [J]. ACS Catal, 2014, 4(3): 833-842.
[43] Jakoblinnert A, Mladenov R, Paul A, et al. Asymmetric reduction of ketones with recombinantE.coliwhole cells in neat substrates [J]. Chem Commun, 2011, 47(44): 12230-12232.
[收稿2016-02-12;修回2016-03-15]
(编辑:谭秀荣)
Advances in bioreduction of imines to chiral amines
ChenYongzheng
(School of Pharmacy, Zunyi Medical University, Zunyi Guizhou 563099,China)
[Abstract]Recently, catalytic asymmetric reduction of imines to chiral amines, mainly including metal catalysis, organocatalysis and biocatalysis, has increasingly become a hot topic in organic chemistry. In this review, we focused on recent progress of the bioreduction of imines to chiral amines involving the screening of imine reductase, related genes cloning and expression as well as the artificial metalloenzyme used for the reduction of imines.
[Key words]biocatalysis; asymmetric reduction; imine reductase; artificial metalloenzyme
[中图法分类号]Q814
[文献标志码]A
[文章编号]1000-2715(2016)02-0107-07
[通信作者]陈永正,男,博士,教授,硕士生导师。主要从事微生物酶的筛选、发现和在手性药物合成中的应用研究,探索化学药物的绿色制造和环境友好合成技术。教育部新世纪优秀人才支持计划获得者、第二批贵州省高层次创新型人才“百”层次入选者、第九批贵州省优秀青年科技人才培养对象、第七批贵州省科技创新人才团队领衔人、第一批贵州省教育厅优秀科研创新团队领衔人、贵州省药学特色重点学科手性药物的化学生物学方向带头人、贵州省仿制药物工程研究中心负责人。现受邀为Organic Letters等12种SCI期刊杂志评议人。主持国家自然科学基金项目3项,在国际知名SCI期刊Angew Chem Int Ed,Adv Synth Catal和J Org Chem等杂志上发表论文18篇,其中2篇论文被著名评论期刊SYNFACTS作为亮点工作进行评价。E-mail:yzchen@zmc.edu.cn。
[基金项目]国家自然科学基金资助项目(NO:21162047);贵州省科技厅基金资助项目(NO:QKHGZ-2011-7017)。
