氧化还原引发体系的宏观动力学研究
2016-05-21刘振国刘嘉渝单振国张春英金春玉
张 涛,刘振国,刘嘉渝,单振国,杨 博,张春英,王 萍,金春玉,董 颖
(1.中国石油吉林石化公司 商务管理处,吉林 吉林132021;2.中国石油吉林石化公司 研究院,吉林 吉林 132021;3.中国石油吉林石化公司 建修公司,吉林 吉林 132021)
过氧化物与亚铁离子的氧化还原体系由于其活化能较低、引发剂分解速率高以及可在低温下聚合等特点,其在工业上被广泛应用于商品胶乳产品及合成树脂的制备,如丁苯橡胶和丙烯腈-丁二烯-苯乙烯共聚物(ABS)等。其具有降低丁苯胶乳的聚合温度,降低1,2加成百分比,减少顺式结构,增多反式结构,并降低胶乳的支化度和交联度等优点,因此引起了人们广泛的研究兴趣[1-10]。虽然其已在工业生产中应用日趋广泛,但其引发机理、其各组分的量及配比等因素对聚合反应动力学的影响还鲜有报道,缺乏完整的了解。
由于丁苯胶乳中丁二烯单体的特殊性(易燃易爆,高压液态存放),对研究其聚合动力学带来极大不便,因此本文以丙烯酸丁酯(BA)为研究对象,采用异丙苯过氧化氢/硫酸亚铁/乙二胺四乙酸钠盐/甲醛次合硫酸氢钠(CHP/FES/EDTA/SFS)氧化还原体系引发其种子乳液聚合。系统研究了其引发机理、各组分用量及配比对聚合反应动力学的影响,确定了各组分的适宜配比。
1 实验部分
1.1 原料
BA:聚合级,需用质量分数为10%的氢氧化钠水溶液洗涤3次后用去离子水洗至中性,加入无水氯化钙干燥后待用;乙二胺钠盐(EDTA)、CHP、 十二烷基硫酸钠(SDS):均为工业级;SFS、N-N二乙基羟氨(DEHA)、硫酸亚铁(FES):均为化学纯。以上原料均由中国石油吉林石化公司提供。实验中所采用的水均为去离子水。
1.2 聚合过程
在装有搅拌器的三口瓶中,加入一定量的去离子水和乳化剂SDS,待其充分溶解后,取预先制备的一定量种子乳液置入该三口瓶中。体系温度恒定为40 ℃,调整搅拌转速为400 r/min,加入一定量BA。向体系通氮气(N2),前10 min将N2进气量适当调大以保证充分除去体系内的氧,再加入配制好的含有水、EDTA、FES、SFS的活化溶液,最后加入氧化剂CHP。反应开始,进行种子乳液聚合制备聚丙烯酸丁酯(PBA)弹性体粒子,直到反应完成。该过程采用的聚合配方详见表1和表2。

表1 乳液聚合制备种子胶乳的典型配方
1) 种子乳液的粒径和固含量分别为110 nm和24%(质量分数);2) 氧化还原引发体系的具体配方详见表2。
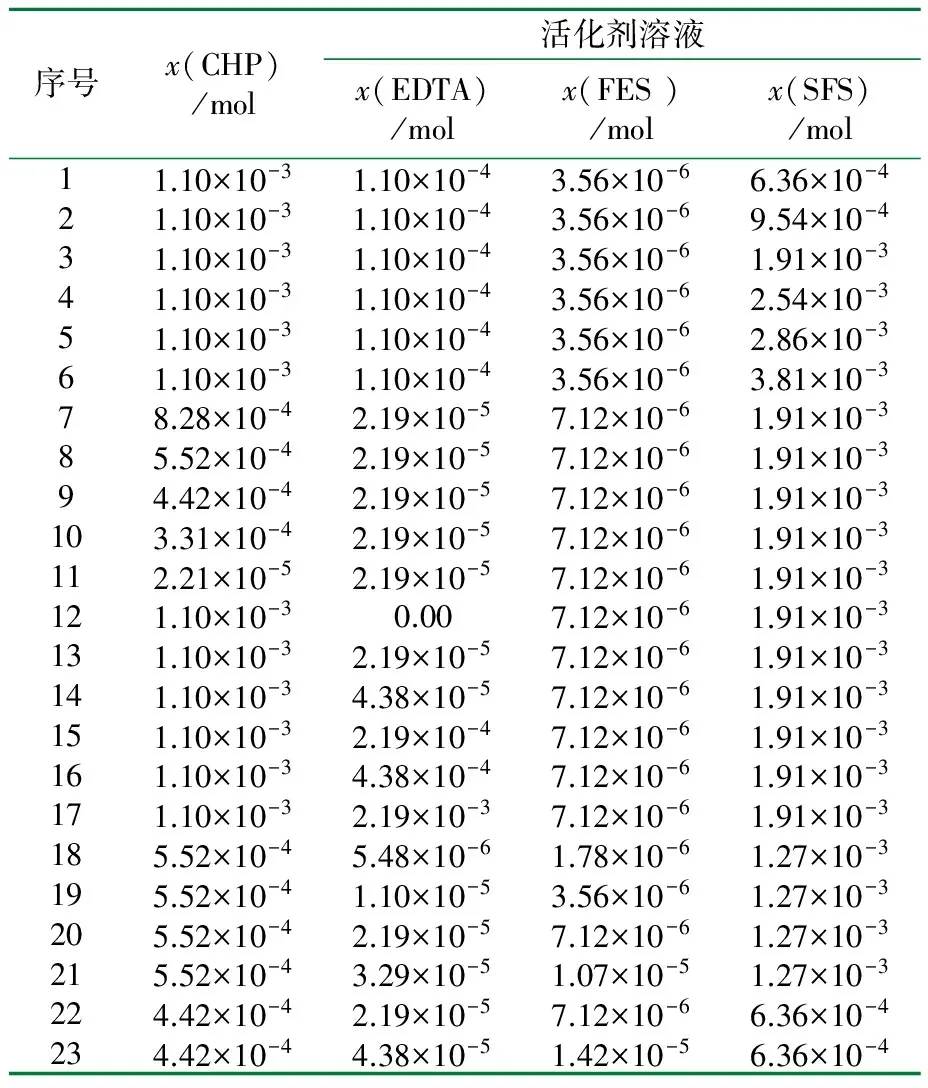
表2 种子乳液聚合中的氧化还原体系的主要配方
1.3 测试方法
1.3.1 动力学研究
反应过程中单体的转化率由质量法测定。从加入引发剂开始计时,在聚合过程中,利用取样器从三口瓶中定期取样,将样品置于铝箔中并加入0.1 mL阻聚剂DEHA以终止反应,并于真空烘箱中100 ℃下烘干至恒重,将所测得的转化率对聚合反应时间作图。
1.3.2 乳胶粒子粒径
本实验所得乳胶粒径使用Brookhaven公司的90Plus粒度仪测定。所测样品需预先利用去离子水稀释至质量分数小于1×10-3%,防止产生由于聚合物粒子中残余单体导致的误差。每个样品在温度为25 ℃、角度为90°条件下,至少测试5次,取平均值。
1.3.3 pH值
利用pH-2C精密数字酸度计,在室温下定期测定体系的pH值。
2 结果与讨论
2.1 氧化还原体系引发机理
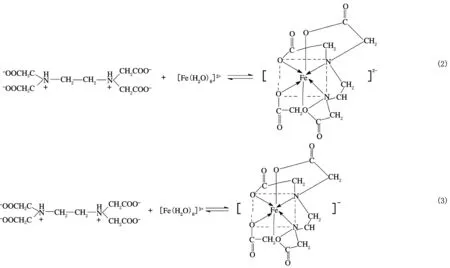
该引发体系中的氧化剂(CHP)为疏水性化合物、主还原剂和助还原剂分别为亲水性的FES和SFS及亲水性的络合剂EDTA。具体反应方程式见式(1)~式(9)。

(1)
(4)
(5)
(6)
(7)
(8)
(9)
在本引发体系中,CHP和Fe2+发生氧化还原反应[见式(1)]生成疏水性的自由基(RO·)。EDTA与Fe2+和Fe3+发生络合反应,形成EDTA-Fe2+络合物和EDTA-Fe3+络合物,控制铁和亚铁离子的释放速率,有效控制反应速率,防止发生爆聚,如式(2)和式(3)所示。SFS与Fe3+反应将其还原成Fe2+[见式(4)],从而使Fe2+可以循环使用,有效降低Fe2+的用量。式(5)~式(9)为体系中其余副反应。
Wall 和Swoboda[11]研究了乳液聚合中的氧化还原引发循环。他们指出,其所采用的乳化剂硬质酸钠不仅起乳化单体的作用,同时与亚铁盐反应生成油溶性盐。如果亚铁离子不能充分溶于油相中,此氧化还原循环将会破坏;还原剂在乳化剂作用下具有油溶性,是此循环能够顺利进行的必要条件,此循环的反应场所为油相;但Wang等在利用氧化还原引发体系CHP/SFS/EDTA/FES引发苯乙烯的微乳液聚合中指出氧化还原引发体系的反应场所是在水油二相界面[4]25-15。本研究中采用SDS为乳化剂,相比于硬质酸钠,其与亚铁盐的反应产物并不具有油溶性,反应能够顺利进行,因此能够证明氧化还原引发体系的引发场所为水油两相界面。图1给出了该引发体系的引发机理示意图。
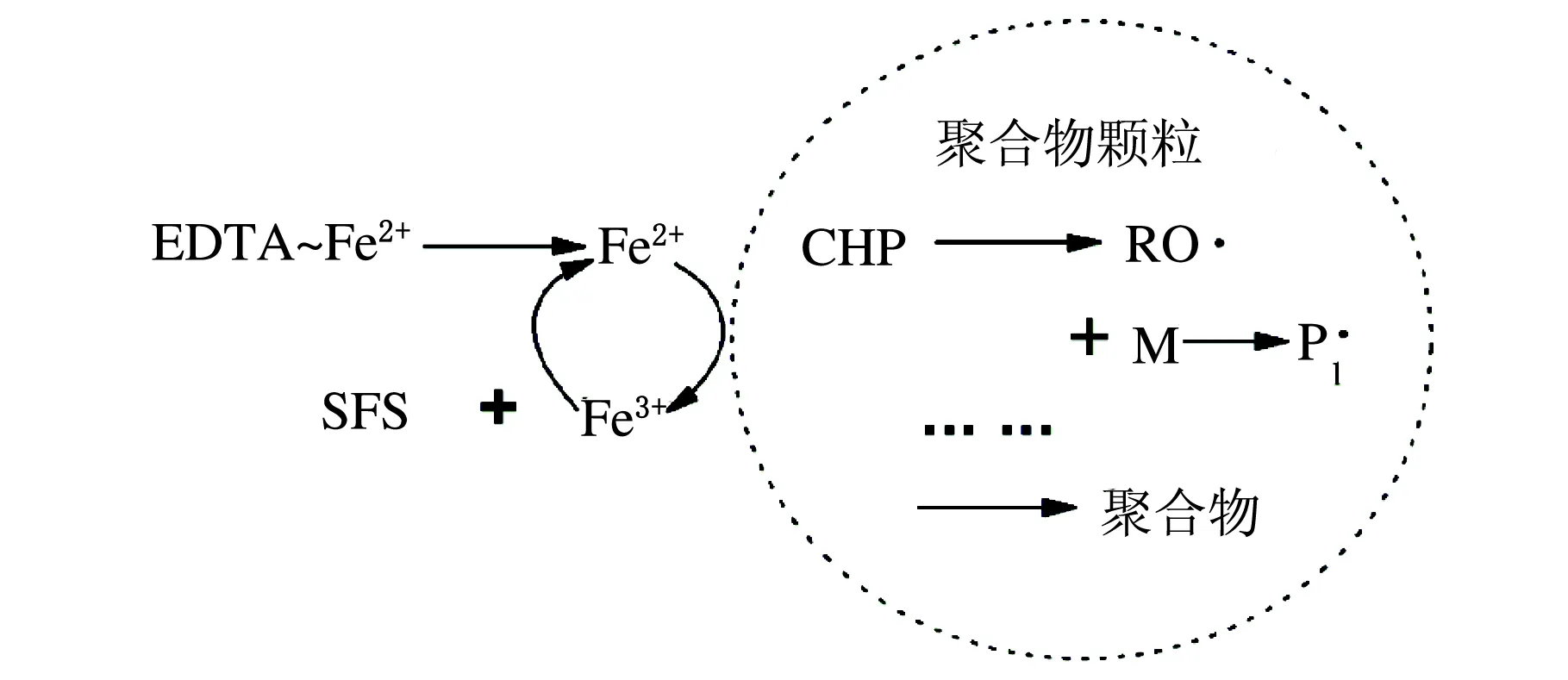
图1 引发过程示意图
从图1可以看到,CHP与Fe2+生成的疏水性的自由基(RO·)首先在聚合物粒子的表面引发BA单体生成低聚物自由基(P1·),低聚物自由基(P1·)扩散至乳胶粒子的内部,继续引发聚合。由CHP和Fe2+反应生成的Fe3+离子可以从粒子表面扩散到水相,与助催化剂SFS相遇而被SFS还原成Fe2+。Fe2+循环地扩散至聚合物粒子表面与CHP继续反应,直到体系中CHP或SFS消耗完毕,反应终止。
2.2 SFS用量对聚合动力学的影响
此引发体系的主反应为CHP与SFS反应生成初始自由基和酸离子,而亚铁粒子仅仅充当二者之间的有效媒介。因此,SFS的用量对于整个反应体系至关重要。SFS可以将高价的铁离子(Fe3+)还原成低价态的Fe2+粒子,从而形成氧化还原循环以保证反应的继续进行[见式(4)]。另外,SFS的加入可以有效避免CHP与Fe3+[见式(8)]以及RO·与Fe2+[见式(9)]之间的副反应。由于温度25 ℃、离子强度约为0时的Fe3+的络合常数为lgKFe3+Y24.3,明显大于25 ℃、离子强度约为0时Fe2+的络合常数lgKFe2+Y14.33[12],如果氧化还原体系中SFS的量少,SFS不能快速有效地将Fe3+还原成Fe2+,大量的Fe3+粒子将会取代Fe2+与络合剂EDTA络合形成更稳定的络合物,导致参加氧化还原循环的Fe2+逐渐减少,反应速率逐渐降低。
实验中恒定EDTA量为1.10×10-4mol,FES量为3.56×10-6mol,CHP量为1.10×10-3mol,考察SFS用量对宏观动力学的影响。图2给出SFS用量对聚合反应动力学的影响。
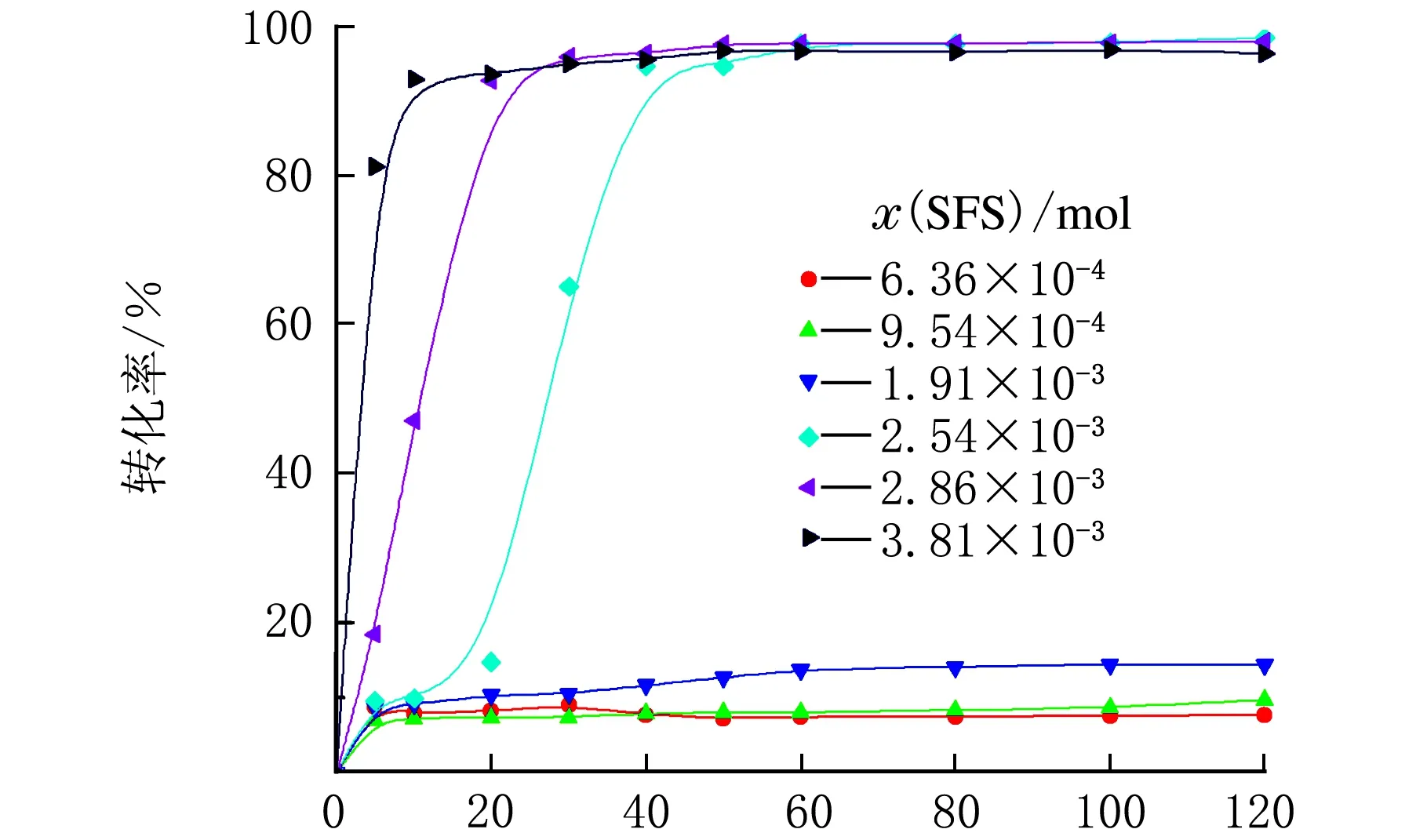
t/min图2 SFS用量对转化率的影响示意图(表2中序号1-6)
由图2可知,随着SFS用量的增加,聚合速率明显增大。但单纯使用CHP/SFS引发体系,虽有反应式(7)发生,但SFS的用量增加对转化率无明显影响。人们普遍认为在氧化还原引发体系中氧化剂与还原剂最佳配比应为等物质的量比[13]。有趣的是,在本研究中,当CHP/SFS的物质的量比为1.10/0.954(近似等物质的量比)时,单体的最终转化率极低(小于20%)。但当CHP/SFS的物质的量比为1.10/3.81时,转化率近乎100%。因此,研究发现,CHP/SFS的最佳物质的量比并非等物质的量比,而是近似为1/3。
另外,实验考察了在这一系列中当SFS量为2.54×10-3mol时的pH变化规律。如图3所示,体系的pH随着聚合反应的进行而逐渐降低。反应式(6)是导致此现象的主要原因。HOCH2SO3Na随着反应而逐渐增加,促使其电离平衡往右移动,不断生成H+,从而导致pH不断降低。
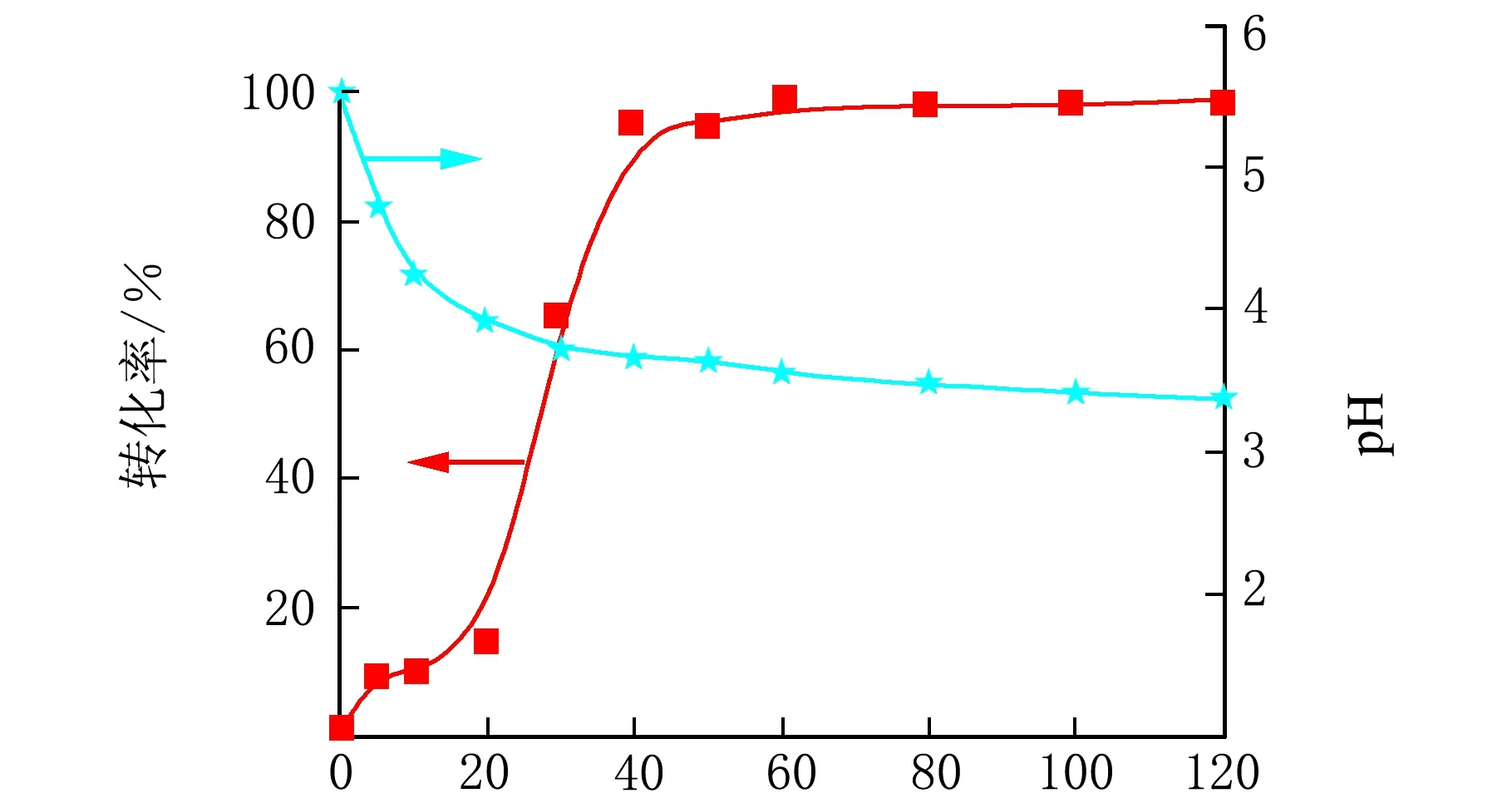
t/min图3 体系pH与转化率的关系示意图(表2中序号4)
Wang[4]2511在苯乙烯的微乳液聚合研究中发现一种现象:苯乙烯的转化率极低(≤20%),即使加入更多的SFS,转化率也没有明显变化,并将之称为“极限转化率”,并指出“极限转化率”是由于在聚合物外层为“玻璃态的苯乙烯”,其阻碍单体和自由基的相遇从而无法聚合。
图4中“A”点时,补加1.27×10-3mol SFS;图中“B”点时,补加EDTA/FES/SFS的物质的量比为2.19×10-5mol、7.12×10-6mol、1.91×10-3mol。
在本研究中(表2中序号22),当EDTA/FES/SFS/CHP的量分别为(mol)2.19×10-5、7.12×10-6、6.36×10-4、4.24×10-4时,也发现了类似情况。如图4所示,在120 min中,单体的总体转化率非常低(<20%)。为了确定是否因为SFS的量不足而导致出现“极限转化率”,向反应体系中补加1.27×10-3mol SFS(图4中“A”点)。从图4中“B”点可以看出,补加SFS对单体的转化率并没有明显影响。但是当继续补加活化剂溶液(EDTA/FES/SFS物质的量分别为2.19×10-5mol、7.12×10-6mol、1.91×10-3mol)后,观察发现单体的总体转化率迅速由15%增加至90%。同时对PBA粒子的粒径变化规律进行了观察,前120 min聚合物粒径无明显变化,仅由110 nm增加至120 nm。然后补加SFS(1.27×10-3mol)仍对聚合物粒径无明显影响。但当补加活化剂溶液后,PBA粒子粒径由144 nm迅速增加至258 nm,这与转化率变化相一致。上述这些现象均表明“极限转化率”产生的原因并非由于“玻璃态聚合物”存在所导致的。
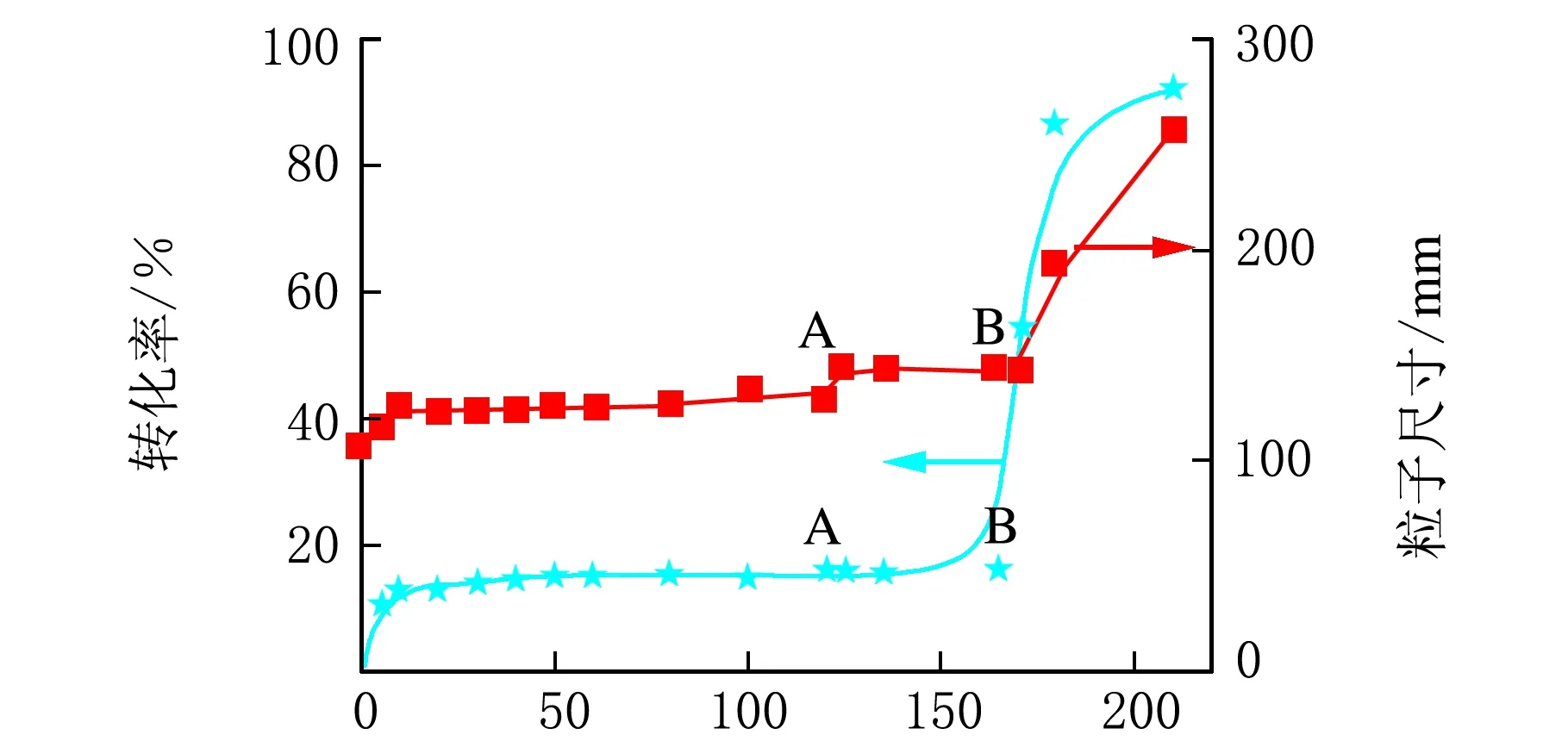
t/min图4 粒径与转化率之间的关系示意图(表2中序号22)
对“极限转化率”产生原因进行推断,其可能是由于 SFS的起始加入量过少,CHP与Fe2+反应生成的Fe3+不能被有效、快速地还原,铁离子与SFS形成稳定的络合物而导致的。这推论可由实际的络合常数lgKMY′证实。实际络合常数lgKMY′受不同pH值影响明显,lgKMY′可由式(10)计算得出:
lgKMY′= lgKMY-lgαY(H)
(10)
式中:lgKMY′、lgKMY和 lgαY(H)分别代表实际的络合常数、络合常数、酸度系数。根据聚合120 min后体系的pH和酸度系数的实测值,分别为3.4、9.70,计算可得lgKFe3+Y′和lgKFe2+Y′分别为14.53、4.63。上述数值表明EDTA-Fe3+络合物的稳定性要明显高于络合物EDTA-Fe2+。
由于EDTA-Fe3+络合物的稳定性较好,体系中游离的Fe3+离子较少,即使再补加SFS也无法形成氧化还原循环;SFS只会与CHP之间发生氧化还原反应,生成一对自由基。而自由基聚合的特点决定两个自由基各自引发聚合的几率远远小于其偶合终止的几率,因此转化率变化不大。
综上所述,由于SFS与Fe3+之间发生氧化还原反应的速率要远远大于Fe3+与EDTA之间的络合速率。一旦CHP与FES反应生成Fe3+,Fe3+将会迅速与SFS发生反应生成Fe2+,而非与EDTA络合。但只要络合物EDTA-Fe3+生成,补加SFS对单体的转化率将无明显影响。也就是说,SFS的初始加入量对于此氧化还原体系起到至关重要的作用。
2.3 CHP用量对聚合动力学的影响
如图5所示,随着CHP用量的增加,聚合速率和单体的总体转化率随之增加。Wang等在苯乙烯的微乳液聚合中发现,聚合反应速率与CHP用量的0.75次方成正比,并且指出即使当CHP的量较高时,也未发现凝胶效应[4]2511。但本研究中,由于采用种子乳液聚合工艺,在种子乳液聚合中,乳胶粒可以长到较普通乳液聚合的乳胶粒大得多,这样由于自由基碰撞时间相对变慢,在同一个乳胶粒中就可能存在几个自由基,最终导致自加速现象,即产生凝胶效应。

t/min图5 CHP用量与单体转化率的关系示意图(表2中序号 7~11)
2.4 EDTA用量对聚合动力学的影响
EDTA和硫酸亚铁可以形成如反应式(2)所示的络合物。EDTA-Fe2+络合物可以通过控制Fe2+的释放速率,即体系中游离的Fe2+浓度,实现对聚合反应速率的有效控制,进而保证聚合反应的平稳进行。EDTA-Fe2+络合物也可以有效避免Fe(OH)3沉淀的生成。如体系中不存在EDTA,过量的二价铁离子就会扩散至界面与氧化剂CHP反应,这样就会导致爆聚,无法保证反应的稳定性。因此,研究EDTA浓度对聚合反应速率的影响具有重要意义。
另外,EDTA-Fe2+的络合方式受体系中的pH值明显影响,在不同pH值条件下,EDTA与Fe2+的络合方式可由Y4-变化到H6Y2+。根据实验观察发现,随着反应的进行,体系酸度变大,因此反应式(2)和式(3)只是其络合方式中的Y4-。
图6给出EDTA用量对聚合反应动力学的影响(表2中序号12~17)。由图6可知,当引发体系中没有EDTA的时候,初始聚合反应速度非常快,转化率在仅仅10 min内就能达到90%。随着EDTA量的增加,当EDTA用量为2.19×10-5mol时(表2中序号13),初始反应引发速率首先增加,单体的总体转化率在5 min内可达90%以上。进一步增加EDTA用量,初始反应速率降低。但EDTA用量对单体的最终转化率并无太明显的影响。换言之,在本研究中,EDTA的用量对整个引发体系并非起到决定性作用,尽管当EDTA/FES物质的量分别为2.19×10-5mol、7.12×10-6mol时初始聚合速率最高。
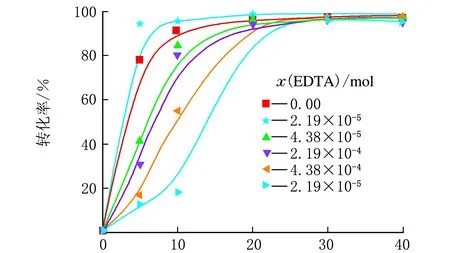
t/min图6 EDTA用量与转化率的关系示意图(表2中序号12~17)
2.5 FES用量对聚合动力学的影响
为了避免EDTA与FES用量变化而导致二者络合方式改变,在本系列中,固定EDTA/FES 物质的量比为 2.19×10-5/7.12×10-6,考察FES用量对聚合动力学的影响。图7给出了硫酸亚铁用量对聚合反应动力学的影响(表2中序号18~21)。
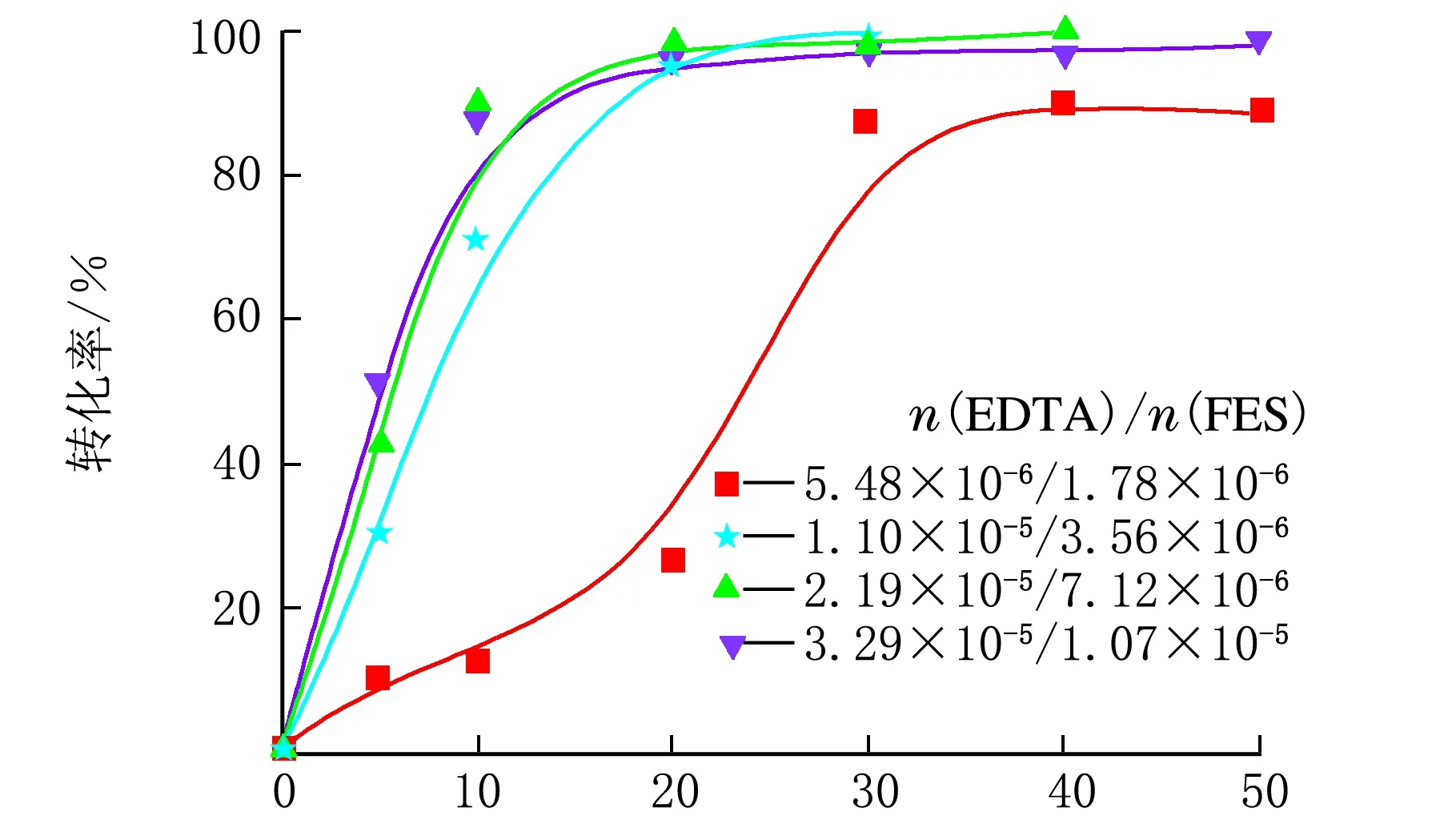
t/min图7 硫酸亚铁用量与转化率关系示意图(表2中序号18~21)
由图7可知,随着FES用量的增加,聚合反应速率明显增加。但当FES用量达到2.19×10-5mol后,进一步增加FES用量对聚合反应速率和单体的最终转化率并无明显影响。
3 结 论
(1) 引发体系的引发机理为CHP与FES通过扩散在界面引发,此引发体系的主反应为CHP与SFS反应生成初始自由基和酸离子,而亚铁离子仅仅充当二者之间的有效媒介。
(2) CHP与SFS最佳物质的量比并非为传统认知的等物质的量比而是近乎1/3。
(3) 聚合反应速率随着CHP和SFS用量的增加而增加;随着EDTA用量的增加,聚合反应速率先增加后减小。
(4) EDTA用量对单体的最终转化率影响不大,EDTA/FES的最佳物质的量比为2.19×10-5/7.12×10-6;但过多FES的用量对聚合反应速率和单体的转化率影响不大。
(5) 通过对“极限转化率”现象研究发现,SFS的初始加入量在此引发体系中起到至关重要的作用。
参 考 文 献:
[1] WARSON H.Redox polymerization in emulsion.In Emulsion Polymerization [M].Washington DC:American Chemical Society,1976:228.
[2] E R Weidlein.Synthetic rubber research in Germany[J].Chem Eng News,1946,36(9):415.
[3] KOLTHOFF I M,MEDALIA A I.Redox recipes:II.Redox recipes in alkaline medium initiated by the system cumene hydroperoxide-iron-sugar at 30 ℃[J].J Polym Sci,1950,5(4):391.
[4] WANG C C,YU N S,CHEN C Y.Kinetic study of the mini-emulsion polymerization of styrene[J].Polymer,1996,37(12):2509-2515.
[5] KOLTHOFF I M,MEDALIA A I.Redox recipes:I.Reaction between ferrous iron and peroxides.general considerations[J].J Polym Sci,1949,4(3):377.
[6] KOLTHOFF I M,MEDALIA A I.Redox recipes:III.Use of various sugars at 0° and 30 ℃ in a cumene hydroperoxide-iron-sugar recipe[J].J Polym Sci 1951,6(1):93.
[7] KOLTHOFF I M,MEDALIA A I.Redox recipes: IV.Dihydroxyacetone recipes at 0 ℃[J].J Polym Sci,1951,6(2):189.
[8] BOLLAND J H.Kinetic studies in the chemistry of rubber and related materials:I.The Thermal oxidation of ethyl linoleate[J].Proc Roy Soc,1946,186(1005):218.
[9] LAITINEN H A,NELSON J S.Determination of hydroperoxides in rubber and synthetic polymers[J].Ind Eng Chem Anal,1946,18(7):422.
[10] LEA C H.The determination of the peroxide value of edible fats and oils:the influence of atmospheric oxygen in the chapman and mcfarlane method[J].J Soc Chem Ind,1945,64(4):106.
[11] WALL F T,SWOBODA T J.An oxidation-reduction cycle in emulsion polymerization systems[J].J Am Chem Soc,1949,71(3):919.
[12] DEAN J A.Electrolytes,electromotive force and chemical equilibrium.In Lange′s handbook of chemistry:Fifteenth edition [M].New York: McGraw-Hill,1999:93.
[13] KOHUT-SVELKO N,PIRRI R,ASUA J M.Redox initiator systems for emulsion polymerization of acrylates[J].J Polym Sci:Part A,2009,47(11):2917.
