鬼臼毒素脲类衍生物的合成和体外抗癌活性的研究
2016-05-18李楠赵阳魏菲高晨光曹波高颖陈虹
李楠,赵阳,魏菲,高晨光,曹波,高颖,陈虹
(1.天津医科大学药学院,天津市临床药物关键技术重点实验室,天津300070;2.天津中医药大学研究生院,天津300193;3.中国人民武装警察部队后勤学院生药与药剂学教研室,天津300309)
论著
鬼臼毒素脲类衍生物的合成和体外抗癌活性的研究
李楠1,3,赵阳1,3,魏菲2,高晨光3,曹波3,高颖3,陈虹1,3
(1.天津医科大学药学院,天津市临床药物关键技术重点实验室,天津300070;2.天津中医药大学研究生院,天津300193;3.中国人民武装警察部队后勤学院生药与药剂学教研室,天津300309)
目的:为了得到抗癌活性更高的抗肿瘤药物,设计、合成新型脲类鬼臼毒素衍生物。方法:以鬼臼毒素或4’-去甲基鬼臼毒素为起始原料经N,N’-羰基二咪唑(CDI)活化其C-4位氨基,并与仲胺类化合物进行缩合反应,得到鬼臼毒素脲类衍生物,经氢谱和高分辨质谱对其进行结构表征。采用噻唑蓝(MTT)法测试目标化合物对人白血病(K562)和人宫颈癌(Hela)细胞的体外抗肿瘤活性。结果:合成8个新的目标化合物,其中化合物6a增殖抑制活性较强。结论:在鬼臼毒素C-4位引入哌嗪环脲类结构可提高其体外抗肿瘤活性。
鬼臼毒素;脲;N,N’-羰基二咪唑;抗肿瘤活性
鬼臼毒素存在于小檗科(Berberidaceae)多年生草本类群鬼臼亚科(Podophylloideae)、山荷叶属(Diphylleia Michx)、八角莲属(Dysosma Woodson)、足叶草属(Podophyllum L.)和桃儿七属(Sinopodophyllum Ying)植物的根和茎中[1]。鬼臼毒素的抗肿瘤作用得到广泛关注,研究者对鬼臼毒素进行结构改造并研制出依托泊苷和替尼泊苷等临床一线抗肿瘤药物,其它衍生物如NK-611、GL331等也进入药物临床前研究[2]。鬼臼毒素类衍生物抗肿瘤作用机制分为两种:一是抑制微管蛋白的组装并妨碍中期相细胞分裂,二是通过抑制拓扑异构酶Ⅱ抑制癌症细胞DNA复制[3-4]。临床中依托泊苷和替尼泊苷等鬼臼毒素衍生物用于治疗睾丸癌、小细胞肺癌和白血病等肿瘤疾病[5]。虽然已合成鬼臼毒素类衍生物具有一定的抗肿瘤活性,却存在骨髓抑制、胃肠道反应、水溶性差等问题[6]。本文在对鬼臼毒素类衍生物活性构效关系研究的基础上,结合文献中对鬼臼毒素的C-4位改造的抗癌活性较好的报道[7-10],运用药物拼合原理,将结构简单、水溶性好的脲类结构与鬼臼毒素结合,设计合成了8个衍生物,对其抗肿瘤作用进行初步探讨。
1 材料与方法
1.1 仪器和试剂 XT显微熔点仪;高分辨质谱仪:maXis ESI-Q-TOF (Bruker Daltonics);核磁共振仪: BrukerAdvance2B/400M;酶标仪:美国,BIORAD550;98%纯度的鬼臼毒素购自上海金和生物技术有限公司;CDI和所用仲胺均从百灵威科技有限公司订购;所用化学合成溶剂均为市售的分析纯或化学纯。VP-16:江苏恒瑞医药股份有限公司,产品批号:09090731;RPMI1640培养基(Gibco公司产品);MTT、胰蛋白酶(均为Sigma公司产品);细胞株:人宫颈癌细胞Hela由中国医学科学院协和医科大学药物研究所药理室陈晓光教授组传代保种。人慢性髓性白血病急变期细胞K562购于中国医学科学院血液学研究所。
1.2 合成方法
1.2.1 合成路线 见图1。

图1 化合物5a-5g,6a的合成路线Fig 1 Schematic route of 5a-5g,6a compounds
1.2.2 中间体化合物3的合成 将鬼臼毒素(414 mg,1 mmol)或4’-去甲基鬼臼毒素(400 mg,1 mmol)溶于20 mL无水二氯甲烷中,小心加入NaN3(260 mg,4 mmol)搅拌使其溶解,冰浴下将1 mL CF3COOH缓慢滴加到反应液中,冰浴下反应1 h,常温反应4 h。用NaHCO3调节pH为中性,分出有机层,无水Na2SO4干燥,减压蒸干,溶于10 mL乙酸乙酯中,加入10%Pd/C(100 mg),HCOONH4(252 mg,4 mmol),加热回流搅拌5 h,用漏斗抽滤,取滤液并用饱和氯化钠水溶液洗3遍,减压蒸干得白色固体即化合物3,其中R1=CH3或H。
1.2.3 化合物5a的合成 取化合物3,其中R1= CH3,即4β-氨基-4-脱氧表鬼臼毒素(103.5 mg,0.25 mmol)溶于无水二氯甲烷(10 mL)中,滴加少量无水三乙胺(0.20 mL),待鬼臼毒素全部溶解后加入N,N’-羰基二咪唑(59.0 mg,0.36 mmol)常温搅拌8 h,得到化合物4,其中R1=CH3,加入无水二乙胺(80.0 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5a。
1.2.4 化合物5b的合成 取化合物4(126.8 mg, 0.25 mmol),其中R1=CH3,溶解于二氯甲烷(10 mL)中,加入无水二正丙胺(111.1 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5b。
1.2.5 化合物5c的合成 取化合物4(126.8 mg, 0.25 mmol),其中R1=CH3,溶解于二氯甲烷(10 mL)中,加入无水N-甲基哌嗪(110.1 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5c。
1.2.6 化合物5d的合成 取化合物4(126.8 mg, 0.25 mmol),其中R1=CH3,溶解于二氯甲烷(10 mL)中,加入无水N-乙基哌嗪(125.5 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5d。
1.2.7 化合物5e的合成 取化合物4(126.8 mg, 0.25 mmol),其中R1=CH3,溶解于二氯甲烷(10 mL)中,加入无水1-(2-吡啶基)哌嗪(179.4 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5e。
1.2.8 化合物5f的合成 取化合物4(126.8 mg, 0.25 mmol),其中R1=CH3,溶解于二氯甲烷(10 mL)中,加入无水N-苯基哌嗪(178.3 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5f。
1.2.9 化合物5g的合成 取化合物4(126.8 mg, 0.25 mmol),其中R1=CH3,溶解于二氯甲烷(10 mL)中,加入无水1-(4-甲氧基苯基)哌嗪(211.3 mg,1.10 mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物5 g。
1.2.10 化合物6a的合成 取化合物3,其中R1= H,即4β-氨基-4’-去甲基-4-脱氧表鬼臼毒素(100.0 mg,0.25 mmol)溶于无水二氯甲烷(10 mL)中,滴加少量无水三乙胺(0.20 mL),待鬼臼毒素全部溶解后加入N,N’-羰基二咪唑(59.0 mg,0.36 mmol)常温搅拌8h,加入无水N-甲基哌嗪(80.0mg,1.10mmol)常温搅拌过夜。依次用体积分数13%盐酸水溶液、饱和碳酸氢钠水溶液、饱和氯化钠水溶液洗涤反应所得液,无水硫酸钠干燥过夜,过滤,滤液减压蒸干,经硅胶柱层析分离提纯,减压蒸干得化合物6a。
1.3 MTT法检测化合物体外抗肿瘤活性 取对数生长期的Hela及K562细胞接种于96孔培养板内,每孔约含4 000个细胞,置37℃,5%CO2细胞培养箱中培养。24 h后给药组加入待测药物,每组设3个平行孔,阴性对照组加入与给药组等体积的溶剂,共孵育48 h后弃培养液,每孔加50 μL 1 mg/mL MTT的磷酸盐缓冲液(PBS)溶液,37℃孵育4 h,弃上清,每孔加入150 μL DMSO溶解甲臜颗粒,轻度振荡溶解。以酶标仪490 nm下测定光密度值,用下面公式计算化合物对细胞生长的抑制率,并以SPSS13.0统计软件计算IC50值,试验平行3次。
抑制率=(阴性对照组OD值-加药组OD值)/(阴性对照组OD值-空白对照组OD值)×100%
2 结果
2.1 化合物的表征数据
5a:白色固体,产量455.9 mg,产率89%,m.p. 156℃-157℃;1H NMR (300 MHz,CDCl3)δ 6.81(s, 1H,H-5),6.47 (s,1H,H-8),6.28 (s,2H,H-2′,6′), 5.96(d,J=2.4 Hz,2H,H-OCH2O),5.12 (d,J=3.4 Hz,1H,H-4),4.54(s,2H,H-1,H-NH),4.45-4.36(m, 1H,H-11),3.89(t,J=9.9 Hz,1H,H-11),3.78(s,3H, H-4′),3.74(s,6H,H-3′,5′),3.26(ddt,J=31.9,14.6, 7.3 Hz,4H,H-N′CH2),2.92(d,J=3.3 Hz,2H,H-2, 3),1.14(t,J=7.1 Hz,6H,H-N′CH2CH3);HRESI-MS:m/z 535.204 8 for [M+Na]+(calcd for C27H32N2NaO8+,535.205 1)。
5b:白色固体,产量470.0 mg,产率87%,m.p. 158℃-159℃;1H NMR (400 MHz,CDCl3)δ 6.80(s, 1H,H-5),6.49(s,1H,H-8),6.29(s,2H,H-2′,6′), 5.97(s,2H,H-OCH2O),5.14(d,J=1.8 Hz,1H,H-4), 4.59-4.53(m,1H,H-NH),4.47(d,J=5.8 Hz,1H, H-1),4.45-4.38(m,1H,H-11),3.93-3.84(m,1H,H-11),3.79(s,3H,H-4′),3.75(s,6H,H-3′,5′),3.30-3.04(m,4H,H-N′CH2),2.91(d,J=2.0 Hz,2H,H-2, 3),1.63-1.48(m,4H,H-N′CH2CH2),0.90(t,J=7.4 Hz,6H,H-N″CH2CH2CH3);HR-ESI-MS:m/z 541.2558 for[M+H]+(calcd for C29H37N2O8+,541.254 4)。
5c:白色固体,产量485.3 mg,产率90%,m.p.161℃-162℃;1H NMR (400 MHz,CDCl3)δ 6.81(s, 1H,H-5),6.51(s,1H,H-8),6.28(s,2H,H-2′,6′), 5.98(d,J=2.6 Hz,2H,H-OCH2O),5.15(dd,J=5.9, 4.4 Hz,1H,H-4),4.74(s,1H,H-NH),4.57(d,J=4.8 Hz,1H,H-1),4.43 (dd,J=9.1,7.3 Hz,1H,H-11), 3.97-3.84(m,1H,H-11),3.80(s,3H,H-4′),3.75(s, 6H,H-3′,5′),3.61-3.44(m,4H,H-N′CH2),3.05-2.88(m,2H,H-2,3),2.57(s,4H,H-N″CH2),2.43(s, 3H,H-N″CH3);HR-ESI-MS:m/z 562.215 6 for[M+ Na]+(calcd for C28H33N3NaO8+,562.216 0)。
5d:白色固体,产量459.2 mg,产率83%,m.p.163℃-164℃;1H NMR (300 MHz,CDCl3)δ 6.79(s, 1H,H-5),6.49(s,1H,H-8),6.26(s,2H,H-2′,6′), 5.96(d,J=3.3 Hz,2H,H-OCH2O),5.14(d,J=2.2 Hz,1H,H-4),4.57(t,J=5.3 Hz,2H,H-1,NH),4.48-4.36(m,1H,H-11),3.95-3.85(m,1H,H-11),3.79(s, 3H,H-4′),3.74(s,6H,H-3′,5′),3.50-3.30(m,4H, H-N′CH2),2.92 (d,J=2.1 Hz,2H,H-2,3),2.53-2.38(m,6H,H-N″CH2),1.11(t,J=7.2 Hz,3H,N″-CH2CH3);HR-ESI-MS:m/z 576.231 5 for[M+Na]+(calcd for C29H35N3NaO8+,576.231 6)。
5e:白色固体,产量481.8 mg,产率80%,m.p.166℃-167℃;1H NMR(400 MHz,CDCl3)δ 8.16(dd, J=4.9,1.6 Hz,1H,H-3″),7.52(ddd,J=8.8,7.3,1.9 Hz,1H,H-5″),6.83(s,1H,H-5),6.71-6.59(m,2H, H-5″),6.49(s,1H,H-8),6.28(s,2H,H-2′,6′),5.97 (d,J=3.4 Hz,2H,H-OCH2O),5.18(dd,J=5.9,3.5 Hz,1H,H-4),4.79(d,J=6.1 Hz,1H,H-NH),4.55(d, J=3.8 Hz,1H,H-1),4.47-4.37(m,1H,H-11),3.91 (dd,J=12.3,7.6 Hz,1H,H-11),3.78 (s,3H,H-4′), 3.74(s,6H,H-3′,5′),3.59(d,J=5.3 Hz,4H,H-N″H2),3.56-3.47(m,4H,H-N′H2),2.95(d,J=3.3 Hz, 2H,H-2,3);HR-ESI-MS:m/z 603.246 1 for[M+Na]+(calcd for C32H35N4O8+,603.244 9)。
5f:白色固体,产量511.1 mg,产率85%,m.p.167℃-168℃;1H NMR(400 MHz,CDCl3)δ 7.28(dd, J=8.7,7.3 Hz,2H,H-3″,5″-Ar),6.91(d,J=8.7 Hz, 3H,H-2″,4″,5″-Ar),6.84(s,1H,H-5),6.50(s,1H, H-8),6.28(s,2H,H-2′,6′),5.96(dd,J=6.3,1.1 Hz, 2H,H-OCH2O),5.18(dd,J=6.0,3.6 Hz,1H,H-4), 4.84(d,J=6.2 Hz,1H,H-NH),4.56(d,J=4.0 Hz, 1H,H-1),4.49-4.37(m,1H,H-11),3.97-3.87(m,1H, H-11),3.78(s,3H,H-4′),3.74(s,6H,H-3′,5′),3.54 (d,J=5.1 Hz,4H,H-N′CH2),3.17(t,J=5.1 Hz,4H, H-N″CH2),2.95(d,J=4.0 Hz,2H,H-2,3);HR-ESIMS:m/z 624.231 4 for[M+Na]+(calcd for C33H35N3NaO8+, 624.231 6)。
5g:白色固体,产量530.3 mg,产率87%,m.p.164℃-165℃;1H NMR(400 MHz,CDCl3)δ 7.82(d,J =8.9 Hz,2H,H-3″-Ar),6.89-6.76(m,3H,H-5,2″-Ar),6.44(s,1H,H-8),6.27(s,2H,H-2′,6′),5.93(dd, J=14.3,0.9 Hz,2H,H-OCH2O),5.29(d,J=6.5 Hz, 1H,H-4),5.21(dd,J=6.3,4.4 Hz,1H,H-NH),4.48 (d,J=4.8 Hz,1H,H-1),4.43(dd,J=9.0,7.4 Hz,1H, H-11),3.97-3.89(m,1H,H-11),3.76(s,3H,H-4′), 3.73(s,6H,H-3′,5′),3.64-3.56(m,4H,H-N′CH2), 3.43-3.32(m,4H,H-N″CH2),3.07-2.83(m,2H,H-2, 3),2.46(s,3H,H-COCH3);HR-ESI-MS:m/z 644.284 9 for[M+H]+(calcd for C33H38N3O9+,644.260 3)。
6a:白色固体,产量472.7 mg,产率90%,m.p.153℃-154℃;1H NMR (400 MHz,CDCl3)δ 6.81(s, 1H,H-5),6.50(s,1H,H-8),6.29(s,2H,H-2′,6′), 5.97(d,J=4.6 Hz,2H,H-OCH2O),5.15(d,J=2.4 Hz,1H,H-4),4.71(d,J=6.0 Hz,1H,H-NH),4.54(d, J=3.5 Hz,1H,H-1),4.45-4.37(m,1H,H-11),3.96-3.85(m,1H,H-11),3.77(s,6H,H-3′,5′),3.52-3.33 (m,4H,H-N′CH2),2.92(d,J=1.9 Hz,2H,H-2,3), 2.44(t,J=5.0 Hz,4H,H-N″CH2),2.34(s,3H,H-N″CH3);HR-ESI-MS:m/z 548.201 0 for[M+Na]+(calcd for C27H31N3NaO8+,548.200 3)。
2.2 化合物体外抗肿瘤活性测定 表1是合成化合物与阳性对照药依托泊苷(VP-16)对K562和Hela细胞株的药理活性结果,可以初步看出8个目标化合物显示出不同程度的对两种肿瘤细胞株生长抑制作用,且部分化合物活性优于VP-16活性。
表1 测试化合物对Hela和K562细胞增殖的抑制活性(s,n=3)Tab 1 Inhibitory activity of target compounds against Hela cell and K562 proliferation(s,n=3)
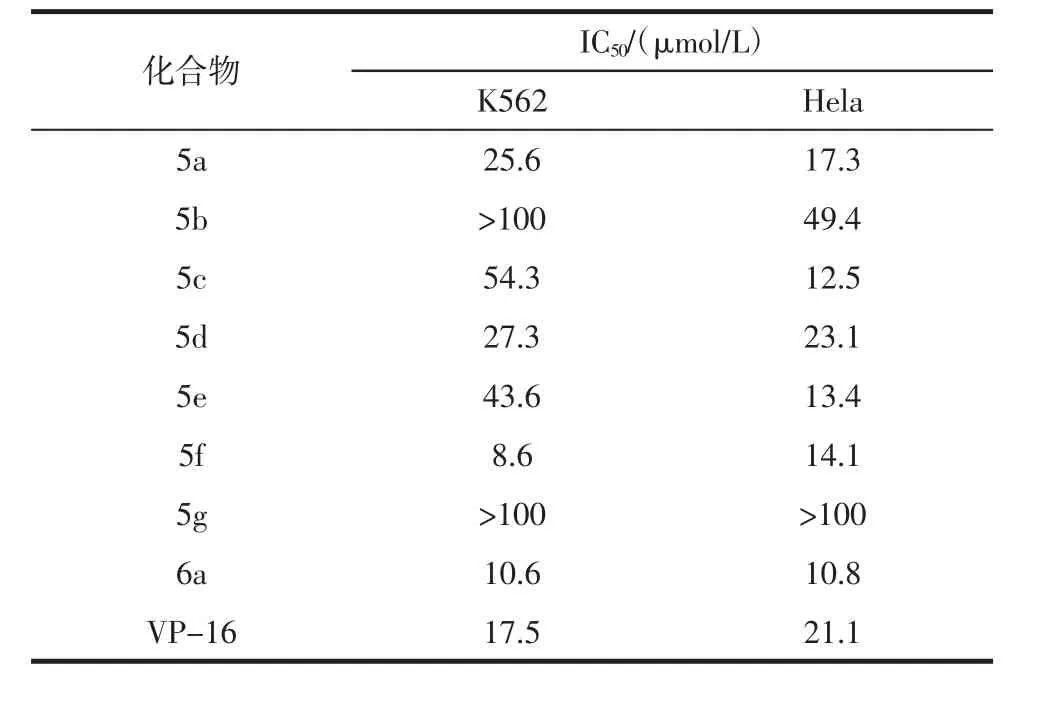
表1 测试化合物对Hela和K562细胞增殖的抑制活性(s,n=3)Tab 1 Inhibitory activity of target compounds against Hela cell and K562 proliferation(s,n=3)
?
3 讨论
3.1 合成方法 本文采用CDI缩合法合成脲类鬼臼毒素衍生物,相比于其他脲类化合物的合成方法,如异氰酸酯法、三光气法、氯甲酸酯法,CDI缩合法合成步骤少、处理反应简单、合成产率相对较高。缺点是对试剂和反应底物的无水要求较高,CDI自身在空气中容易吸潮失效,不易把握用量标准。反应中所接基团不宜太大,否则空间位阻影响产率和产物纯度;中间体化合物4不稳定容易分解,应及时监测反应进度。
3.2 构效关系 VP-16又名依托泊苷,拓扑异构酶II抑制剂,临床上应用于小细胞肺癌的一线化疗药物,以VP-16作为阳性药是由于其在临床上的广泛应用,以及其基本母核与鬼臼毒素相关,能够说明改造后的效果是否显著。根据表1所示药理结果可以看出,化合物5 b和化合物5 g抑制肿瘤细胞增殖作用较差,推测是正丙胺基和甲氧基的引入降低了抗肿瘤活性。其中6 a和5 f的体外抗肿瘤活性较好,提示哌嗪环的引入利于提高鬼臼毒素抗肿瘤活性,且其位阻相对适当,引入苯环等芳香环状结构将降低衍生物的抗肿瘤活性,化合物6 a活性最好,具有进一步研究前景。
[1]杨显志,邵华,张玲琪.鬼臼毒素资源研究现状[J].中草药,2001, 23(11):85
[2] Liu X,Zhang L L,Xu X H,et al.Synthesis and anticancer activity of dichloroplatinum(II)complexes of podophyllotoxin[J].Bioorg Med Chem Lett,2013,23(13):3780
[3] Choi J Y,Cho H J,Hwang S G,et al.Podophyllotoxin acetate enhances γ-ionizing radiation-induced apoptotic cell death by stimulating the ROS/p38/caspase pathway[J].Biomed Pharmacother, 2015,70:111
[4] Kamal A,Telukutla S,Polepalli S,et al.Synthesis and biological evaluation of 4-aza-2,3-dihydropyridophenanthrolines as tubulin polymerization inhibitors[J].Bioorg Med Chem Lett,2014,24(15): 3356
[5] Liu J F,Sang C Y,Xu X H,et al.Synthesis and cytotoxic activity on human cancer cells of carbamate derivatives of 4β-(1,2,3-triazol-1-yl)podophyllotoxin[J].Eur J Med Chem,2013,(64):621
[6] Kamal A,Suresh P,Ramaiah M J,et al.4β-[4′-(1-(Aryl)ureido) benzamide]podophyllotoxins as DNA topoisomerase I and IIα inhibitors and apoptosis inducing agents[J].Bioorg Med Chem, 2013,21(17):5198
[7] Zhang Z J,Tian J,Wang L T,et al.Design,synthesis and cytotoxic activity of novel sulfonylurea derivatives of podophyllotoxin[J]. Bioorg Med Chem,2014,22(1):204
[8] Ai T,Shi S Y,Chen L T,et al.Synthesis and anti-tumor activity evaluation of novel podophyllotoxin derivatives[J].Chinese Chemical Letters,2013,24(1):37
[9]Cheng W H,Cao B,Shang H,et al.Synthesis and evaluation of novel podophyllotoxin derivatives as potential antitumor agents[J].Eur J Med Chem,2014,85:498
[10]Zhang Z H,Zhang L M,Luo G,et al.Synthesis and biological evaluation of novel podophyllotoxin analogs as antitumor agents[J].J Asian Nat Prod Res,2014,16(5):527
(2015-10-08收稿)
Synthesis and biological evaluation of 4-ureido podophyllotoxin derivatives as anticancer agents
LI Nan1,3,ZHAO Yang1,3,WEI Fei2,GAO Chen-guang3,CAO Bo3,GAO Ying3,CHEN Hong1,3
(1.School of Pharmacy,Tianjin Medical University,Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics,Tianjin 300070,China;2.Graduate School,Tianjin University of Traditional Chinese Medicine,Tianjin 300193,China;3.Department of Pharmacognosy,Logistic University of PAP,Tianjin 300309,China)
Objective:To obtain anticancer agents with higher efficiency through a novel series of 4-ureido podophyllotoxin derivatives.Methods:The novel target compounds were synthesized by reacting podophyllotoxin or 4’-demethylepipodophyllotoxin with secondary amines under catalytic agent N,N’-carbonyldiimidazole(CDI).All the compounds were characterized by1H-NMR and HR-MS. Meanwhile,MTT assay was used to test their cytotoxicity against K562 and Hela cell lines.Results:Eight novel target compounds were synthesized.Among them,compound 6a exhibits was identified to have stronger inhibition effect.Conclusion:The anticancer activity of podophyllotoxin could be improved when its C-4 is combined with the structure of ureido and piperazidine.
podophyllotoxins;urea;N,N’-carbonyldiimidazole;antitumor activity
R9
A
1006-8147(2016)03-0199-05
国家自然科学基金资助项目(30873363);天津市应用基础项目资助(08JCYBJC070000)
李楠(1990-),男,硕士在读,研究方向:天然产物结构改造;通信作者:陈虹,E-mail:chenhongtian06@163.com。
