第五章 三大合成催化
2016-05-17孙文华
孙文华
第五章三大合成催化
孙文华
(中国科学院化学研究所,北京 100190)
5.1引言
5.1.1合成高分子的历史发展
伴随人类出现,就开始了使用天然高分子材料的历史,包括使用树木和植物搭建窝棚及帮助攀爬等,在进化过程中开始使用藤蔓植物和芦苇、树皮和棉花等的编织物,直到19世纪中叶,开始跨入天然聚合物的化学改性时代。1839年Charles Goodyear使用硫化反应使天然橡胶更具弹性和实用性[1];1865年John Wesley Hyatt使用樟脑增塑硝化纤维素[2],并于1870年实现了硝化纤维塑料的工业化。20世纪进入合成高分子时代,1907年Leo Henricus Arthur Baekeland首次实现了热固性酚醛树脂的合成[3],成为20世纪20年代实现工业化的第一个合成塑料。开启高分子科学研究,公认是Hermann Standinger教授1920年定义高分子是由结构单元重复并通过普通共价键彼此连接而形成的长链分子,奠定了高分子科学发展的基础。合成高分子先驱Wallace Hume Carothers 1930年4月与合作者Arnold M Collins使用分离的氯丁二烯做单体合成了氯丁橡胶,被认为是第一种合成橡胶;不仅如此,1934年Wallace Hume Carothers集中开展纤维的合成研究,利用二胺和二酸进行缩合反应制备了聚酰胺,基于此,与Gerard Berchet合作于1935年2月使用1,6-己二胺与己二酸反应制备了6,6-聚酰胺(颇具政治色彩的是由于6,6-聚酰胺能够制备高弹纤维,展示了蚕丝的功能,在20世纪30年代,日本是蚕丝制品最大生产国。Wallace Hume Carothers所在的DuPont公司期望6,6-聚酰胺的纤维能够实现对于日本蚕丝的替代,采用“Now You Lose Old Nippon”的缩写Nylon命名了6,6-聚酰胺)。Wallace Hume Carothers的研究充分展示了合成高分子的两类重要反应:加成反应与缩合反应。与此同时,由于德国Standinger H教授(1932年)大分子长链结构理论的确立和前苏联谢苗诺夫H H的链式聚合理论(1934年)的提出,为加成高分子合成奠定了科学基础。英国帝国化学工业公司的Fawcett E W在英国皇家化学会Faraday会议上报告了乙烯高压聚合制备聚乙烯[4],该技术于1939年实现了百吨产业化生产;当时拥有相关技术的还有DuPont和UCC公司,聚乙烯材料生产和应用还仅仅作为军用物资,仅在第二次世界大战后获得更多公司推广。量变的过程必然带来质的跨越,20世纪50年代初,德国科学家Karl Ziegler发展了钛配位乙烯(及烯烃)聚合催化剂,意大利科学家Giulio Natta快速推进了钛配位丙烯聚合及产业化。
20世纪50年代末开始的大规模聚烯烃产业化,使得价廉质优的高分子材料更具民用价值和意义,更为广泛的高聚物研究与应用成为当时最具发展潜力的学科之一。目前合成高分子的塑料、合成橡胶和合成纤维使用总量超过3亿吨,成为提高人类生活水平与减缓地球资源消耗的重要保障材料,成为人们衣、食、住、行和工农业生产必要的物资。
5.1.2合成高分子材料的重要价值
工农业生产与现代服务业发展的目标是提高人类生活水平;不仅如此,为了保障后代的繁衍生息,需要减缓天然资源的使用与保护环境,求得持续与长久发展。尽管民众错觉认为“合成高分子材料是环境污染的罪魁祸首”,设想一下没有合成高分子材料所提供的支持和保障,维持目前人类三分之一人口生活水平的天然物质基础会显得异常困难,地球资源的消耗和温室效应都会呈级数演变。
伴随人类和动物从穴居到窝棚和住宅的进化,快速地消耗着石材和树木,造成森林快速减少,甚至成片消失;空气中温室气体浓度加大,加速了冰川消融和海平面的升高,慢慢吞噬着人类长期居住的家园。这类环境的破环,才真正无法再生。与之对应,高分子材料被冠以“环境的公敌”,却在当今建筑材料中发挥着越来越重要的作用,提供了轻量化、高强度、隔热、抗冲击和抗震的优良材料,已经成为临时建筑的框架与墙体和固定建筑中防渗漏、涂料、密封剂、黏合剂、给排水、电讯、隔音与保暖等不可缺少的材料,更是快速发展的城市中高层建筑必须的轻量建材。科学总是“双刃剑”,高分子建材和家装材料存在易燃与老化及事故中有毒烟雾的缺点备受批评,其替代钢铁、水泥与木材的功能,直接保护了自然资源和提高了人类的居住环境与生活品质。材料科学家仍然在使用掺杂和共混持续地提高高分子材料的性能和克服其存在的缺点。
比居住条件更为重要的是“衣”,人类通过天然纤维的应用提高了审美和生活的品质,这些天然纤维包括植物纤维的棉与麻以及动物纤维的毛与蚕丝等。在地球人口数量超过60亿的当下,天然纤维已经远远无法满足量的供给,更无法满足人们对于衣物的色泽、保暖、坚固耐磨、防霉防蛀和免洗免烫等特点的追求;即使热恋天然织物的人,也不难发现自己的“棉、麻、毛和蚕丝”衣物很难占到两成,甚至很有可能标记为“天然纤维”的衣服中仍然含有合成高分子材料。不仅如此,高分子纤维鲜艳的光泽和美感备受时尚界的青睐和更具商业价值;例如:尼龙长袜具有棉质感和更具保暖与易洗涤的腈纶休闲服装,透气性能与干爽效果更佳的维尼纶内衣织品等,无疑撼动了天然高分子纤维的特征价值。除高分子纤维之外,装点普通百姓衣物的还需要饰品与纽扣,其基本原料更多出自高分子合成材料。市面上流行的仿珍珠纽扣与饰品取材于聚酯,仿玉制品基于聚酯或者聚甲醛树脂,这些饰品有更光滑的表面和靓丽的色泽,甚至可以比较便利地包裹小昆虫与植物做成琥珀饰品,给人更高档的色感和冲击力。值得补充说明,高分子纤维的衣物和饰品使用更具耐久性,没有高分子纤维对于衣物和饰品的基石,天然材料无法满足现在人类对于衣物的欲望需求的十分之一。
与人类生活品质密切相关的还有“行”,高分子材料在交通工具部件和资源节约中的价值更是毋庸置疑。包括汽车车体与内外加护和装饰材料,飞机、轮船和火车的框架与部件以及内外装饰材料。对于钢铁与木材的替代,以及高分子材料轻量化带来的动力消耗大幅的减少,都对自然的保护起了巨大作用!尽管在部分产品中暴露了强度不足的安全性和燃烧性问题,碳纤维材料无论在汽车和飞机甚至空间卫星制造中都成为强度高和耐久与耐热性能俱佳的材料。高分子材料使用的范围与性能要求可以满足制造不同层次交通工具的需求,人们不能都采取最高材料标准的关键还是材料制备的工艺与技术过程的能耗问题。无疑,使用更多高分子材料制造交通工具成为材料科学与制造业发展的主流。
不仅如此,如果说“高分子材料甚至提高了你的饮食质量”,你会如何反应?高分子材料并非用于食用,但是在食品储运与保鲜中高分子材料的作用至关重要,隔断了细菌传播和滋生的条件,甚至极大地减少了交叉感染的可能,保护了身体,提高了生活质量,满足了人类需求。大家在争论一次性餐具和哪种做法更保护自然环境。拿一个常被诟病的“一次性塑料杯和陶瓷杯”招待客人哪个更环保进行比较,即使不计算清洗和用水,制备一个陶瓷杯的能耗是一次性塑料杯能耗的两千倍!至于其环境的负面影响,这些高分子包装和容器完成功能后最有效的利用手段就是焚烧和作为能源的原料;此外,也有再降解成为有机单体和产业化工程装置的研究,显得更具吸引力与持续发展前景。
5.1.3合成高分子材料分类
高分子材料种类众多,学术界和产业界有很多不同的分类方式,尤以按照材料使用时的形态分类被广泛接受,分为“塑料、纤维和橡胶”三大类。毋容置疑,涂料和胶粘剂作为合成高分子材料越来越受到重视,但是在催化科学为主的教材中,这里仅简单介绍“塑料、纤维和橡胶”。
5.1.3.1塑料
塑料又称为树脂,“树脂”顾名思义,是植物渗(泌)出物的无定形(半)固体有机物;天然树脂的使用可以追溯到数千年前,最早使用的天然树脂是松香、虫胶和琥珀等。尽管现代塑料工业大家比较接受的是始于20世纪30年代,但可以追溯到19世纪中叶为了寻找天然树脂的代用品,美国人海厄特J W在湿润的硝酸纤维素中加入樟脑和少量酒精制成了一种可塑性物质,能够热压下成型,命名为“赛璐珞”替代虫胶,用于马车和汽车的挡风玻璃和后来作为电影胶片的材料,并于1872年建厂生产赛璐珞;1903 年德国人艾兴格林A发明了不易燃烧的醋酸纤维素和注射成型方法,1905 年德国拜耳股份公司进行工业生产。与此同时,美国的Albang Dental Plate公司于1870年最早研究酚醛树脂的合成,后由美国Bakelite(酚醛树脂)公司于1909年将该技术实现产业化生产。比美国更早进行第一个工业化酚醛树脂生产的是1902年布卢默(Blumer L)用酒石酸催化酚和醛缩合制得命名为“Laccain”的酚醛树脂,遗憾的是没有形成规模;被称为酚醛树脂创始人的美国科学家巴克兰(Baekeland)是1905年才开始用苯酚和甲醛来合成树脂的研究,并于1909年首次提出酚醛树脂“加压和加热”的热固性塑料,奠定了其科学威望,并在1924年担任美国化学会主席。1910年在柏林吕格斯工厂建成了通用酚醛树脂公司,更加提升了酚醛树脂的批量生产;应该说在20世纪40年代前,酚醛塑料约占塑料产量的三分之二,是最主要的塑料品种,广泛用于电器、仪表、机械和汽车工业。
在跨入现代塑料工业过程中,还值得提及1930年德国法本公司进行工业聚苯乙烯生产,1931年美国罗姆-哈斯公司以本体法生产聚甲基丙烯酸甲酯(有机玻璃);以及20世纪40年代用乳液法生产聚氯乙烯。然而,具有现代塑料工业里程碑的研究是1933年英国卜内门化学工业公司(ICI)两位研究人员Reginald Gibson和Eric Fawcett在进行乙烯与苯甲醛170 ℃加压反应时,发现聚合釜壁上有蜡质固体存在,发明了聚乙烯,并于1939年实现高压气相本体法生产低密度聚乙烯;该技术在第二次世界大战中由多国公司生产聚乙烯,当时聚乙烯还仅仅限于战备物资使用。塑料获得普及和大量应用还是在1953年联邦德国Karl Ziegler(齐格勒)教授领导的实验室发明烷基铝活化四氯化钛常压催化乙烯聚合,并在1955年联邦德国赫斯特公司首先采用低压下制备高密度聚乙烯;受齐格勒研究的启发,意大利塑料改性共混研究的学者纳塔G抓住机会突击丙烯聚合研究,其技术于1957年由意大利蒙特卡蒂尼公司实现了聚丙烯工业生产。此后的发展则一发而不可收,塑料世界总产量从1904年的10 kt,到1956年达到3.4 Mt,2012年使用量超过了220 Mt(见图5-1)。大品种塑料中包括聚烯烃、聚苯乙烯、聚碳酸酯、ABS树酯、聚苯醚和聚酰亚胺等;其中聚烯烃占了塑料总量的近三分之二。

图 5-1 全球塑料产品消费量(2012年超过220 Mt)
5.1.3.2合成橡胶
1860年,威廉斯C G在热裂解天然橡胶产物时收集到了异戊二烯,并且发现异戊二烯在空气中放置会变成白色弹性体,布查德 G又在1879年重复了该技能。1900年孔达科夫И Л采用2,3-二甲基-1,3-丁二烯聚合制备弹性体,2,3-二甲基-1,3-丁二烯在70 ℃热聚合历经 5个月后获得弹性体称为甲基橡胶W,而在(30~35) ℃聚合历经3~4个月后制成略硬的橡胶称为甲基橡胶H。在第一次世界大战期间,德国的海上运输被封锁,切断了天然橡胶的输入,该技术于1917年在德国获得工业化生产;然而,其性能远比天然橡胶差得多,停战后随即停产,仅生产了2.3 kt。前苏联列别捷夫 C B利用酒精转化成的丁二烯采用钠催化液相本体聚合,制得了丁钠橡胶,在1931年建成万吨级生产装置;同一时期,德国人从乙炔出发合成丁二烯,也实现了丁钠橡胶的制备,德国法本公司1935年首先实现丁腈橡胶生产和1937年在布纳化工厂建成丁苯橡胶工业生产装置,而丁腈橡胶是一种耐油橡胶,目前仍是作为特种橡胶使用。第二次世界大战的急需和当时日本占领了天然橡胶主产地马来西亚,促进了橡胶产业的快速发展,世界合成橡胶产量从 1939年的23 kt剧增到1944年的885 kt。不仅如此,还出现了许多特种橡胶新品种,包括美国通用电气公司1944年生产的硅橡胶,同期德国和英国分别生产了聚氨酯橡胶。
随着齐格勒-纳塔烯烃聚合工业化,石油化工裂解制备乙烯与丙烯过程中产生了大量二烯单体,配合新型催化剂研发的溶液聚合技术更有效地控制橡胶分子的立构规整度,提高了橡胶性能,使合成橡胶工业进入一个崭新的阶段。主要品种包括:接近天然橡胶性能被称为合成天然橡胶的高顺式1,4-聚异戊二烯(简称异戊橡胶),高反式1,4-聚异戊二烯(简称合成杜仲胶),顺式1,4-聚丁二烯橡胶(简称顺丁橡胶)以及丁腈橡胶、氯丁橡胶和丁苯橡胶等;此外还有乙烯与丙烯共聚制备的乙丙橡胶和聚氨酯弹性体等。到20世纪70年代后期,合成橡胶已基本可以替代天然橡胶制造各种轮胎和制品,而且特种合成橡胶具有天然橡胶无法达到的性能与品质。目前合成橡胶的年产量约7 Mt。
5.1.3.3合成纤维
在合成高分子中另一个重要分支就是“合成纤维”。1913 年,德国人Klatte F合成了聚氯乙烯纤维,该技术于1934年由德国IG化学公司实现工业化生产,是最早的合成纤维,然而,由于耐热性差阻碍了其应用与发展。同期美国科学家、缩聚合成高分子的奠基人Carothers W H博士于1935年使用己二胺和己二酸缩聚合成了聚酰胺66,DuPont 公司在1938年实现了中试生产,1939年纺丝成功聚酰胺66纤维,俗称尼龙;当1940年投放市场,立刻成为世界上争相抢购的织物,成为第一种大规模生产合成纤维。1941年英国“Calico Printers Association”公司的Whinfield J R和Dickson J T成功地使用对苯二甲酸与乙二醇实现缩聚,并于1944年成功实现熔体纺丝;该技术于1947 年由英国ICI实现产业化生产聚对苯二甲酸乙二醇酯纤维,在1953年DuPont 公司从英国购买了专利进行聚酯纤维“Dacron”的大规模生产。聚酯纤维成本低和用途广泛,至1972 年聚酯纤维产量超过聚酰胺,成为最大量和应用最广的合成纤维。
然而,合成纤维另一个平行的增长在于Ziegler-Natta催化剂制得廉价的聚乙烯和聚丙烯成为新的纺丝材料,意大利Montefibre公司自1960年就实现了聚丙烯纤维的工业化,吸引了国际产业界的竞争。2013年合成纤维84.5 kt,其中聚烯烃纤维占52.7 kt,其他重要合成纤维还有聚酯、聚酰胺、聚丙烯腈等,合成纤维的快速发展也挤压了棉花的生产与销售空间,使天然纤维呈现萎缩局面。
此外,作为一般信息,还需特别提及两种特种纤维:碳纤维和超高分子量聚乙烯纤维。碳纤维质量比铝材轻,强度却高于钢铁,并且在有机溶剂、酸、碱中不溶不胀及耐腐蚀和模量高等特点,成为航空和国防军工的重要物资,并在民用奢侈品和耐用品市场开始使用,呈现出巨大潜力。碳纤维生产主要分为聚丙烯腈基碳纤维、沥青基碳纤维等,其生产具有战略意义,备受航空与军工企业重视;国家多次组织重点攻关和产学研联合研究,实现了丙烯腈聚合和纺纱制备原丝,在将原丝放入氧化炉中300 ℃进行氧化后,再升温到(1 000~2 000) ℃进行碳化工序处理,制备碳纤维,在国内已有吉化公司实现了规模化和批量生产(其他民营公司也采用相似的技术工艺)。超高分子量聚乙烯纤维(Ultra High Molecular Weight Polyethylene Fiber,简称UHMWPE),又称高强高模聚乙烯纤维,是目前世界上比强度和比模量最高的纤维,其分子量在1×106~5×106的聚乙烯冻胶纺丝制成的纤维,具有抗紫外线辐射,防中子和γ射线,比能量吸收高、介电常数低、电磁波透射率高等特点;最初集中用于防弹衣、防弹头盔、军用设施和设备的防弹装甲、航空航天等军事领域,近年来用于缆绳、防切割手套和渔网(养鱼箱)等制备。其技术核心是乙烯聚合中对于超高分子量聚乙烯分子量和分子量分布的有效控制,以及冻胶(干法和湿法)纺丝品质控制。我国在2008年奥运前作为重点攻关项目由原北京东方化工厂助剂二厂(现并入中国石化燕山分公司)联合中国科学院化学所使用中国石化奥达催化剂公司催化剂实现了万吨级产业化,国内多家公司成功用于纺丝和制备相关产品,其防弹衣、防弹头盔与防弹装甲保障国家需求,还出口西亚诸国。
5.2高分子材料合成中典型催化问题
无论现代化工还是高分子材料的合成过程无疑需要催化工艺,降低日益增加的劳动力和场地成本,而且在提高生产效益的同时,改善了产物的品质。在高分子材料合成按照反应类型分为加成聚合和缩聚反应,需要给出必要的实例进行探讨。在上面介绍中不难看出,合成高分子材料基于其功能分为塑料、合成纤维和橡胶。然而,基于科学本质,科学研究关心原料和反应过程以及所得材料的微结构,产物材料的微结构控制了其宏观性能和应用领域与范围。
目前,占据塑料产量市场三分之二和合成纤维中超过六成的都是基于聚烯烃材料,而且聚烯烃微结构调控延伸到橡胶替代的弹性体材料。聚烯烃材料以其“成本低、重量轻和易加工”的特性,而且仅仅限于碳氢化合物的组成保证其使用后仍可作为燃料的环保品质,成为重点推广的材料;不仅如此,聚烯烃树脂生产能力与技术水平也是衡量一个国家石化产业发展水平的重要标志。聚酯其组成限于碳氢氧,其制品在完成使用功能后无论降解还是焚烧的产物都具有环境耐受力,而且广泛用于塑料制品与合成纤维的原材料。橡胶作为不饱和键存在、碳氢组成的高分子材料,具有无法替代的性能和应用。聚烯烃、聚酯和橡胶材料使用量占到目前合成高分子材料的九成,其催化技术无疑主导了高分子材料合成的效益和未来发展趋势,掌握了这三类催化技术,不难展开进行其他高分子材料的催化研究。为此,我们在重点介绍聚烯烃催化的基础上,对聚酯和橡胶催化进行必要阐述。
5.2.1聚烯烃材料及其催化剂
自1939年聚乙烯开始工业化生产以来,聚烯烃的发展至今已有70多年的历史,最初的烯烃采用醇脱水制备;20世纪50年代,石油裂解制备烯烃技术极大地提升了石油化工产品的附加值,更重要的是改善了人们的日常生活,被认为是20世纪最伟大的发现之一。聚烯烃具有优良的材料性能,使用中无毒和稳定性能高;通过共聚与改性可制备特种专用树脂,具有高抗冲、高耐热、高透明、低热封温度、导热、导磁或高屏蔽等性能。虽然聚苯乙烯科学上分类为聚烯烃,但是通常俗称的聚烯烃还是专指聚乙烯和聚丙烯及其共聚物;在石油裂解制备烯烃单体时,总是乙烯产量高于丙烯,因而从20世纪中期聚丙烯为主发展到聚乙烯更受到关注。作为聚烯烃性能与分类的知识,其本质还是微结构“支化度多寡与长短,分子量高低与分布宽窄”影响到宏观指标“密度和熔融指数”与材料性能。以乳白色半透明蜡状固体的聚乙烯为例,分为高密度、中密度、低密度和超低密度聚乙烯。密度在(0.941~0.965) g·cm-3的高密度聚乙烯(HDPE)通常由Ziegler-Natta催化剂或铬系(Phillips)催化剂制备,支链化程度最低,结晶度较高,强度和抗老化性能优于聚丙烯,使用温度优于聚氯乙烯,广泛用于注塑、吹塑、挤塑和旋转成型制品加工;此外,超高分子量聚乙烯(UHMWPE)也是高密度聚乙烯中的一员,其密度甚至接近1.0 g·cm-3,采用Ziegler-Natta催化剂和低温(60 ℃)聚合制备,分子量超过100万,用于制备高强和高模聚乙烯纤维,也用于高强板材制备。密度在(0.916~0.940) g·cm-3的中密度聚乙烯(MDPE),结晶度约75%,拉伸强度较高密度聚乙烯差,其刚性、耐磨性和透气性介于高密度聚乙烯和低密度聚乙烯之间,适于用作挤塑管材、蒸煮袋的内衬薄膜和包装材料等。密度在(0.910~0.925) g·cm-3的低密度聚乙烯(LDPE),通常采用高温和高压自由基聚合制备,常称为高压聚乙烯,分子量一般在50 000~500 000,结晶度较低,具有良好的耐低温性能,可用于(-60~-80) ℃的工作温度,电绝缘性能好,是挤塑管材和电线、电缆的重要原料。密度更低的材料称为线型低密度聚乙烯(LLDPE)以及超低密度聚乙烯(VLDPE),通常由茂金属(metallocene)或Ziegler-Natta催化剂进行乙烯与α-烯烃共聚合制备,由于α-烯烃形成侧链造成结晶度低和密度小,但具有良好的表面光泽性和低温韧性高的特点,其高模量、抗弯曲和耐应力开裂性的优点广泛用于吹膜和包装材料。
我国第一套聚烯烃生产装置是20世纪70年代引进日本三井的聚丙烯成套装置在燕山投产;聚烯烃工业发展模式一直是关键技术依赖进口,因而,买方市场的后果是大量装置满足的仅仅是中低档聚烯烃材料的需求。考虑到国际聚烯烃企业对于我国企业的联合抵制,许多引进聚合技术和装置在引进之初并非先进,造成在我国成为聚烯烃材料生产大国和Ziegler-Natta催化剂重要生产国的当下,消耗量超过四分之一的高性能聚烯烃树脂仍然依赖进口。关键根源在于基础研究发展的薄弱和技术开发的滞后,导致我国缺少自主知识产权的“聚烯烃催化剂和聚合工艺”,因而,迫切需要催化剂和催化工艺上创新研究和技术突破。
5.2.1.1铬系(Phillips)催化剂
负载铬系催化剂是基于硅胶或者二氧化硅与氧化铝复合颗粒作载体负载铬,由于是Phillips公司设在俄克拉荷马州的研究室成员J Paul Hogan和Robert L Banks于1951年的发明[5],并且由Phillips公司于1955年实现连续法工业生产,又称为Phillips催化剂。尽管出于环境保护的需求,国际上对于废除铬使用的呼声很高,该催化剂体系仍然生产国际大量消耗的高密度聚乙烯份额中的四成,其原因在于铬系催化剂所得高密度聚乙烯具有较宽的分子量分布,在满足材料性能要求的同时,具有良好的加工性能,备受成品制造者的青睐。
制备铬系催化剂,通常采用硅胶颗粒悬浮在溶剂中,加入水溶良好的铬酸铵,获得硅球吸附三氧化二铬的颗粒(图5-2)[6],然后高温焙烧制得具有催化聚合活性的铬系催化剂,其中铬在负载催化剂中的负载质量分数为0.2%~2%。

图 5-2 氧化铬在硅胶载体表面的负载
20世纪70年代,六价铬被确认为致癌物(因而,国内尚存部分实验室使用重铬酸钾洗液用于清洗玻璃仪器的做法值得停止),为了减少铬系催化剂制备中操作者与六价铬的接触,纷纷采用三价铬盐替代[7],特别是产业界最常用的醋酸铬,在负载催化剂焙烧过程中很容易将三价铬氧化为六价铬,其阴离子则很容易挥发掉。大量的研究证实,铬离子变价转化比较容易,考虑到乙烯聚合是一个相对强的还原气氛,科学家使用光谱跟踪验证了分别使用一氧化碳和氧气环境中铬的氧化与还原状态,认为获得了六价铬和二价铬的证据(图5-3[6]),其他价态组分中间体存在值得推测。

图 5-3 CO/O2气氛下六价铬与二价铬间的还原与氧化
尽管各种价态的铬都被证实或者准确地讲是猜测为催化乙烯聚合的活性组分,也被用作解释铬系催化剂所得聚乙烯分子量分布较宽的根源。六价铬是桔红色,三价铬是蓝色,实验表明,三氧化铬(六价铬)在超过200 ℃处理时很容易转化为低价铬[8];而实际操作中,工业铬系催化剂使用前都是需要焙烧处理才能充分发挥其催化聚合活性,因而,近些年的研究逐渐接受和确认三价铬为催化聚合的活性中心。最新的研究更加验证了20世纪60年代Cossee提出的催化聚合机理[9]。

图 5-4 乙烯聚合链增长和终止的Cossee机理
该机理认为,乙烯作为还原气氛下,高价铬首先还原成为低价铬,配位吸附的乙烯转化为烷基或者氢在铬上键联,形成催化中间体。基于这个烷基键联在铬上的催化中间体(图5-4),单体乙烯进行配位,发生持续乙烯插入反应形成了长链聚乙烯链;一旦这个聚乙烯链足够长,新的配位乙烯单体与之发生氢迁移,就产生了最终的聚乙烯以及新的烷基联接在铬上的新催化中间体;新的催化中间体可以持续诱导乙烯进行新的聚合反应。尽管也有很多文献报道了 “铬键联烷基”和“铬键联氢”催化中间体的证据,但科技界普遍接受这种结论。这类催化中间体如何形成成为普遍关注的科学问题,毕竟铬系催化剂可以不采用烷基金属助催化剂就可以直接实现乙烯聚合。
一种解释是硅胶中羟基的氢转移到铬上,形成“铬键联氢”的催化中间体,然后配位乙烯和插入反应形成“乙基铬”催化物种,持续进行乙烯单体配位和插入获得聚乙烯(图5-5)[10]。
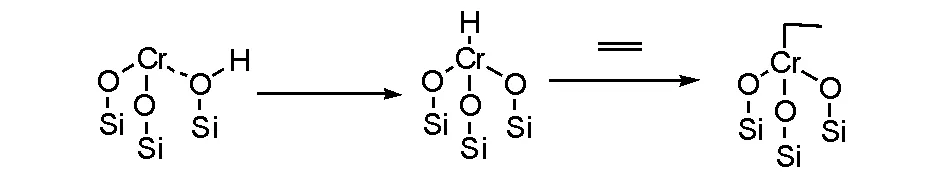
图 5-5 基于硅胶羟基氢引发的链增长
另一种机理解释(图5-6)是基于二价铬活性物种配位乙烯形成的乙叉基铬(乙基卡宾铬化合物),再与乙烯配位插入形成铬杂环丁烷,再进行1,3氢迁移形成烷叉基铬,重复乙烯配位插入,在进行2,3氢迁移时给出含有双键的聚乙烯(或齐聚物)和二价铬活性物种[11]。
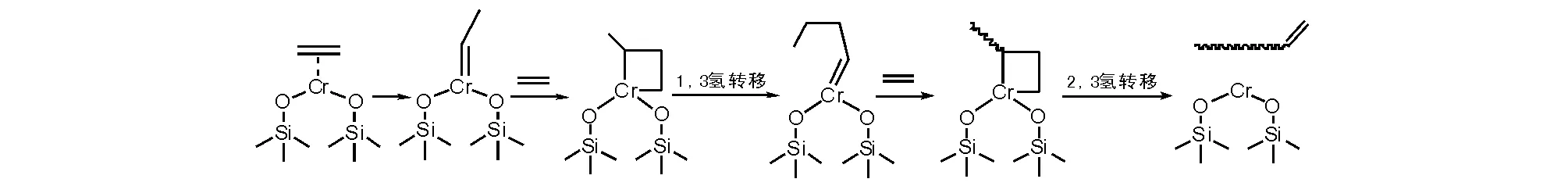
图 5-6 基于二价铬烷叉基乙烯插入成环链增长机理
还有一种机理解释也需要特别关注,就是认为催化过程中形成了铬杂戊烷中间体活性中心(图5-7)[12]。成环机理中给出几个重要信息能够帮助解释铬系催化剂催化乙烯三聚制备己烯:氢迁移形成端烯基中间体以及丙烯基配位的催化中间体,而丙烯基中间体可以转化形成双键位移到内部的烷烃,这也是为什么所得聚乙烯未必能够清晰测试其双键的位置。

图 5-7 基于铬杂环烷基与丙酰基铬催化中间体的机理
基于铬系催化剂实用价值和巨大的商业利益,产业界仍会持续进行改良研究。由于铬潜在致癌的问题,催化剂在聚烯烃中的残留必将影响到该类聚乙烯树脂的使用范围在其被替代前,提高铬系催化剂效率和性能将是永恒的研究主题。同时,仍有值得科学界去解决的谜团,这类基础研究显得更艰巨,无论反应机理和催化中间体的研究,目前计算化学的研究显示了新的途径,能否与实践结果相符以及在什么程度上吻合仍然有待观察。
5.2.1.2钛系(Ziegler-Natta)催化剂
Ziegler-Natta催化剂是以德国科学家Ziegler K和意大利科学家Natta G两人名字命名的最重要石化工业催化剂;自20世纪50年代问世以来,经过不断研究和升级换代,催化性能不断提高,推动了聚烯烃工业的迅猛发展,其生产规模不断扩大,高性能聚烯烃树脂层出不穷。然而,德国人教材中总是将这类催化剂称为Ziegler催化剂;一个金属有机背景的化学家Ziegler和颇具材料性能研究造诣的科学家Giulio Natta却“泾渭分明”地研究这类催化剂对于乙烯和丙烯的聚合,正是两人的重要贡献分享了1963年的诺贝尔化学奖[在Ziegler研究组实现钛催化乙烯聚合之后,同期开始了丙烯催化的研究,遗憾的是Ziegler研究组没能得出丙烯聚合的结果。研究材料性能的Natta教授应邀参加了Ziegler组的催化乙烯聚合研究突破的发布会,会上提问中丙烯聚合研究的进展情况被告知“该催化剂(绝对)不能催化丙烯聚合”的结论触动了Natta的研究兴趣。也许就是“当事者迷,旁观者清”,Natta回到意大利就物色合成和催化的合作者开展研究,在证实Ziegler的乙烯聚合研究结果之后,对于丙烯催化却观察到了蜡状产物的少量产生——丙烯聚合实现了!带着这个结果,Natta又去拜访Ziegler和他的同事,侧面问他们是否再试了丙烯催化;德国人告诉他,不间断地试探并没有获得突破。Natta获得坚强的信心和动力,决定尽快搞清两个催化体系中的不同;但是,Natta仍然没有告诉德国同事意大利学者实现了丙烯聚合。德国人良好的金属有机合成与操作基础保证了“Et3Al-TiCl4”体系纯净,而意大利学者使用反应体系密闭差,造成体系变化最终证实“Et2AlCl-TiCl3”催化丙烯聚合。科学的突破成就了两位科学巨人,然而,也造成两位巨人的个人恩怨,甚至1963年的诺贝尔化学奖颁奖时两位都无法同台,意大利学者在颁奖活动结束后才去领奖。这个故事也提供了创新的例证:新研究人员加入所观察到的离奇研究结果或现象,不应该随便忽视,更应该认真找到原因,也许正孕育一个伟大的发现]。
钛系催化剂的研究起源于1953年德国化学家Ziegler K用Et3Al-TiCl4实现了乙烯的常温、常压聚合,得到了线性的高结晶度聚乙烯,即高密度聚乙烯[13-14]。1954年,意大利化学家Natta G[13]用Et2AlCl-TiCl3催化丙烯聚合,首次得到了全同立构的聚丙烯[15]。理性和客观地讲,Ziegler和Natta两个课题组的工作是相互补充和竞争中获得发展,缺少任何一部分都可能使Ziegler-Natta在聚烯烃工业发展的里程碑推迟。当时Ziegler研究组发现的催化体系,1 g钛只能产生(1.5~3.0) kg聚乙烯,意味着聚乙烯中钛残留300×10-6~1 700 ×10-6;不仅所得聚合物有色,而且影响聚乙烯的稳定性,与使用的铬系催化剂相比活性上没有任何竞争力,而且当时还没有人考虑铬的毒性问题。与之对比,Natta研究组的催化体系受到更多关注,实现了丙烯聚合,由于是获得全同立构的聚丙烯,就具有了快速结晶效果,结晶聚合物体现出更高的强度,当时铬系催化剂无法实现。不仅如此,当时的烯烃(乙烯和丙烯)是通过醇脱水实现,丙烯制备更简单和易于产业化,成为产业界竞相研究和产业化的新材料。
科学研究中相同概念和主题研究间的启发非常关键,钛系催化剂很快借鉴了铬系催化剂负载的概念,使用不同载体[MgCO3,SiO2/Al2O3(当时铬系常用载体),其他金属氯化物和氧化物等]进行氯化钛负载制成的催化剂[16],活性获得极大提高,发展了被称为第二代钛系催化剂(第一代钛催化剂并没有获得应用),进入钛催化乙烯聚合工业化生产阶段。
对于负载钛催化剂催化乙烯聚合的机理,一个趋向是接受Cossee机理(图5-4),显然可以接受。还有一个后来普遍接受被称为Green-Rooney的机理[17-18],该机理与铬系成环催化中间体(图5-6)的机理类似;认为金属活性中心周围有充分的空位进行配位和形成金属氢化物,成环与链增长获得聚乙烯(图5-8)。

图 5-8 乙烯聚合的Green-Rooney机理
同位素标记研究认为,Green-Rooney机理需要进行必要的修正,中间体中有“抓氢键(agostic)”中间体化合物形成,被称为“临位抓氢键参与的乙烯聚合机理”(图5-9)[19]。

图 5-9 临位抓氢键参与的乙烯聚合机理
以上研究主要集中在钛催化剂催化乙烯聚合的方面,源自典型Ziegler催化剂的研究范畴,但是作者无意忽视Natta催化剂使用钛催化剂催化丙烯聚合而获得全同立构的聚丙烯,提高了所得产品聚烯烃的固化速率和强度。在Natta催化剂催化丙烯聚合之前,使用铬系催化剂催化丙烯聚合,获得了无规聚丙烯产品,因材料很难固化而没有受到重视。在丙烯参与聚合时,由于多了一个甲基在双键上,会有1,2-插入和2,1-插入两种聚合的位置选择性插入聚合方式,考虑到甲基的立体选择性问题,会产生四种结构有所差异的聚丙烯产物,如图5-10所示。

图 5-10 丙烯插入聚合的三种方式与所得结构差异的四种聚丙烯
前面提过四价钛和三价钛的氯化物盐用于乙烯聚合和丙烯聚合的区别,而进行催化剂制备的技术人员通常采用催化剂的颜色判断钛的价态:四价钛为白色,负载催化剂固体呈现浅色;三价钛具有暗紫色,其负载催化剂呈现较重的颜色。已经指出过,Natta的丙烯聚合催化剂认为是由“Et2AlCl-TiCl3”红色催化体系组成,在最初十多年聚丙烯生产所需催化剂制备过程中,人们已经习惯使用氢氧化镁和四氯化钛在有乙醇存在下进行反应制备氧化镁负载的三价钛的氯化物,该反应过程中四价钛显然被乙醇还原形成了三价钛化合物。早期的负载催化剂研究,无论乙烯聚合还是丙烯聚合催化剂,都能够看到金属氯化载体对于催化聚合活性的良好贡献。期间,日本三井化学和三菱化学都在与德国和意大利联合开发新的负载催化剂,使用了氯化镁负载钛的催化剂体系,使得催化聚合活性有了质的飞跃。此前的催化聚合所得聚烯烃中会有催化剂残留,钛残留量高于30×10-6时造成聚烯烃树脂有色,氯残留量高于30×10-6时容易降解和腐蚀;因而,第二代Ziegler-Natta催化剂催化聚合所得聚烯烃通常需要洗掉无机盐灰分,该过程通常称为“脱色处理”工序。当氯化镁负载催化剂工业化时,所得聚烯烃树脂中钛和氯的残留量都低于30×10-6,在聚合工艺中免除了“脱色处理”工序;因而,在20世纪60年代末和70年代初研究的这类催化剂被称为“第三代Ziegler-Natta催化剂”, 如对丙烯的聚合活性达到600 kg PP·(g Ti)-1,等规度达到98%[20-21]。
事实上,20世纪70年代后,聚烯烃Ziegler-Natta催化剂的活性提高非常有限,已不是研究的重点。燕山公司引进的我国首套聚烯烃生产的工业装置是日本三井化学的工艺与设备,使用第三代Ziegler-Natta催化剂。针对这个催化剂体系,当时中国科学院化学所和隶属于化工部的北京化工研究院以及石油部的石油科学研究院联合攻关,获得具有实用价值的聚丙烯催化剂,基于该技术分化和扶持了我国三家重要的聚烯烃催化剂生产厂家,分别是奥达催化剂公司、营口向阳化工厂和燕化高新催化剂公司;在国家部委整合情况下,燕化高新催化剂公司并入中国石化属下的奥达催化剂公司,成为国际上颇具影响力和技术领先的聚烯烃催化剂生产企业。此后的40年里,人们把最初重视催化剂活性的研究转到集中和关注所得聚合物树脂的性能。针对聚烯烃树脂的高性能,两个有效途径就是烯烃共聚和立体可控聚合;烯烃共聚的问题异常复杂,我们将在茂金属锆系催化剂部分进行涉及,立体可控聚合对于丙烯聚合更具有实质意义,后续的研究形成了第四代Ziegler-Natta催化剂。在第四代Ziegler-Natta催化剂中,少数催化剂改进通过载体控制实现[22],更多注意力是投到了伴随钛负载催化剂使用的脂类或者醚类化合物,其作用认为是可以调节催化活性中心的电子效应,被称为“给电子体”[23]。基于这类“给电子体”加入到催化剂体系顺序差异,制备载体催化剂时已经引入的脂或醚被称为“内给电子体”,而在催化体系中采用与载体催化剂共混加入的这类脂或醚被称为“外给电子体”。有时,会采用脂或醚同时做内给电子体和外给电子体,也会有多种脂或醚做给电子体;相关研究试探了大量的脂肪族和芳香族脂或醚以及二脂和二醚等。尽管有学者在发展给电子体催化剂体系中提出第五代Ziegler-Natta催化剂的建议,其效果和科学本质仍然是第四代催化剂的范畴。虽然难说Ziegler-Natta催化剂已经是“完美”的,但就催化活性而言,已达到每摩尔钛催化剂每小时实现制备数千吨聚丙烯,全规聚丙烯选择性超过98%的立体规整性。国内重要催化剂研究和生产厂家,中国石化奥达催化剂公司和营口向阳化工厂,都实现了第四代Ziegler-Natta催化剂的生产,在满足国内聚烯烃生产的同时,出口到亚洲甚至欧美。
5.2.1.3锆系(metallocene)催化剂
在讨论锆系催化剂时,不得不回到Ziegler-Natta催化剂的活性中间体研究探索的背景,进而逐渐演化成为锆系(茂金属)催化剂的发现历史。发现Ziegler-Natta催化剂正值金属有机化学学科的建立,聚烯烃催化学者力图通过金属有机化学的方法解释催化机理和活性中间体[24],而金属有机化学家则选择具有重要应用价值的催化体系展示金属有机的普遍性和实用价值[25],研究中一个交叉的重要体系就是二氯二茂钛配合物与烷基铝的催化体系(图5-11)。表现出催化活性的助催化剂是二烷基氯化铝,典型的是氯化二乙基铝进行反应时形成二茂烷基化钛氯化物,进一步与二烷基氯化铝,形成二茂烷基钛正离子催化活性中心[26-27];然而,当两个烷基二茂钛氯化物(如乙基二茂钛氯化物)进行分子间β-氢转移和形成一个乙烷分子脱去后,便产生乙基桥联的氯化二茂钛,该中间体没有适合进行乙烯配位的空间,在进行歧化放出乙烯后形成氯化二茂钛,三价钛失去催化活性[28-29]。

图 5-11 茂金属催化烯烃聚合的活性中间体与失活物种
这类乙基桥联的氯化二茂金属化合物,其锆的化合物获得了单晶,并测定了结构[30],证实乙基桥的每个碳原子同时联到两个锆原子上。与此同时,为更好地使用核磁跟踪催化中心的烷基化过程和研究催化聚合机理,而且为了更好地控制氢转移的速率以便观察茂基钛的催化中间体,使用甲基比乙基更能够简化图谱和便于研究。针对二甲基二茂钛与三甲基铝的反应进行跟踪,成功地分离到甲基中α-氢转移形成的亚甲基桥联的茂基钛化合物[31]。尽管证实该化合物中钛的确以四价态形式存在,但是这类中间体并没有呈现乙烯聚合活性;令人惊奇地发现,核磁管中凝结的水促进了乙烯聚合反应,并直接引入少量水在一升反应釜中再现了这类聚合催化[32]。该结果就是聚烯烃中具有里程碑意义的助催化剂甲基铝氧烷(MAO),40年里,甲基铝氧烷的结构问题一直吸引着研究者的注意,比较普遍接受的观点就是不同聚集态的氧桥联甲基铝簇合物[33]。二茂锆与三甲基铝形成的体系对乙烯和丙烯都没有催化性能,但二茂锆与甲基铝氧烷组成的催化体系却比其同系物二茂钛与甲基铝氧烷的体系无论乙烯还是丙烯聚合都高出两个数量级;不仅如此,茂基锆化合物更容易制备和具有相对较好的溶解性,诱发了茂锆催化剂的探索,引发了茂金属催化烯烃聚合的集中研究与产业化发展,并成为近20年高性能聚烯烃树脂研究与发展的推动力。
茂锆配合物催化剂研究最多的是二茂锆化合物的衍生物,并发展了不同桥联的二茂锆化合物,具有良好端烯烃聚合性能,具有代表性和不同结构对称性模型的二茂锆化合物见图5-12。在合成这些二茂锆配合物过程中,也合成了同族的钛和铪二茂金属配合物,对同等条件下催化乙烯聚合性能进行比较[34],茂锆配合物总是聚合活性较高。虽然茂锆配合物的合成比茂钛配合物容易,但前过渡金属高价态的高亲氧性还是增加了合成的难度,使得合成收率容易受到操作条件的影响。茂锆配合物与Ziegler-Natta催化剂在催化乙烯聚合活性上同在一个数量级,Ziegler-Natta催化剂也可以通过聚合条件变化控制所得聚乙烯的分子量,尽管茂锆配合物催化剂体系可以获得聚乙烯的分子量分布窄,但客观地讲,在乙烯聚合应用上,茂锆催化剂并不占任何优势。
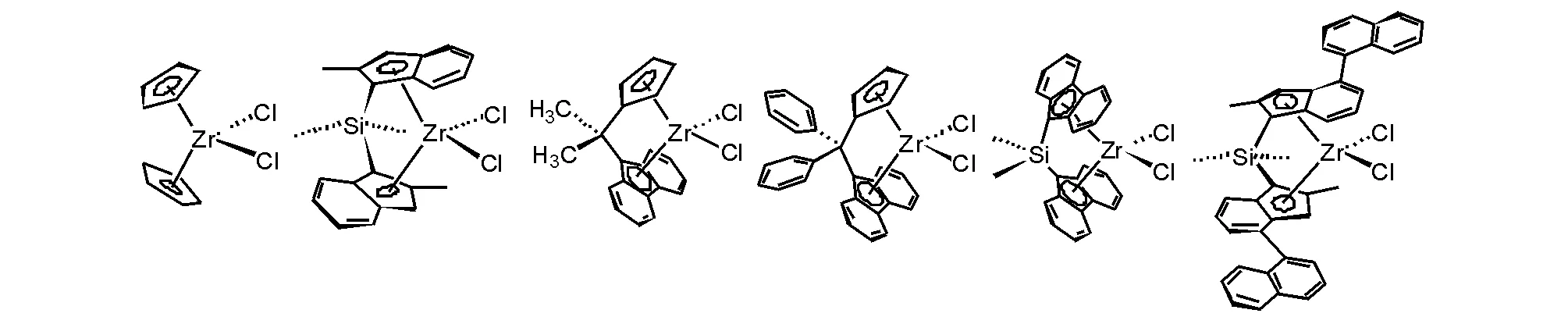
图 5-12 二茂锆催化剂的代表性模型
发挥茂金属催化剂到极致的是其特效性实现可控聚合和共聚合。茂锆催化剂用于α-烯烃的可控聚合能够制备立体规整性不同和性能差异的聚烯烃树脂,如聚丙烯树脂材料(图5-13)。

图 5-13 聚丙烯的不同立体规整结构
聚丙烯材料中甲基的取向造成立体规整性差异,直接影响材料的结晶速率和结晶度以及密度和材料的物理与机械性能;在分子量相同的情况下,等规聚丙烯容易结晶成型,具有良好的耐热性和机械强度,可以用于制造塑料制品和容器;无规聚丙烯耐热性能差,结晶困难,呈现粘稠状或蜡状材料,在改性后使用,例如用于制备改性沥青等。
茂锆催化剂在聚烯烃中最具特殊地位,是催化乙烯与烯烃共聚最有效的催化剂体系,制备了品种繁多的新型高性能聚烯烃树脂。针对聚烯烃树脂材料性能提高进行的烯烃共聚研究是伴随着烯烃自聚出现就诞生的研究,并且实现了钒系催化乙丙共聚胶催化体系仍有使用,Ziegler-Natta催化乙烯与α-烯烃(丁烯-1,己烯-1和辛烯-1等)的共聚是目前工业生产大品种线性低密度聚乙烯(LLDPE)的重要工艺。然而,钒系催化剂催化效率远低于茂金属催化剂体系,呈现茂金属催化剂替代钒系催化剂的局面;在追逐更高机械性能和提高抗撕裂性能的材料研发中,需要提高乙烯与α-烯烃共聚树脂中α-烯烃的共聚比例,不仅如此,还需要长链α-烯烃(如辛烯或者癸烯)的有效共聚,Ziegler-Natta催化剂就难于达到要求,需要茂金属催化剂解决。还有一种特殊的功能聚烯烃树脂,即乙烯与环烯烃(环戊烯或降冰片烯)共聚获得工程塑料型聚烯烃树脂,具有良好的透明性和抗撞击性能。能够实现共聚催化的茂金属催化剂获得学术界广泛关注和研究,也有多个品种实现了产业化应用,但最值得知道和应用的高效茂金属催化剂是硅桥联四甲基茂基叔丁基胺化钛二氯化物(图5-14),即单茂桥联钛配合物催化剂,也被称为“限定几何构型的催化剂”(constrained geometry catalysts,CGC催化剂)。这个催化剂专利权最初归陶氏化学(已过保护期),并与除中国外的世界聚烯烃大公司分享专利使用权,用于生产高性能聚烯烃,多数采用溶液聚合工艺,成为近20年来聚烯烃树脂竞争发展的重要领域。由于国际聚烯烃对于高附加值市场的垄断和我国聚烯烃
工程产业化能力差,我国还没有规模化使用茂金属催化剂制备高性能聚烯烃的装置运行。
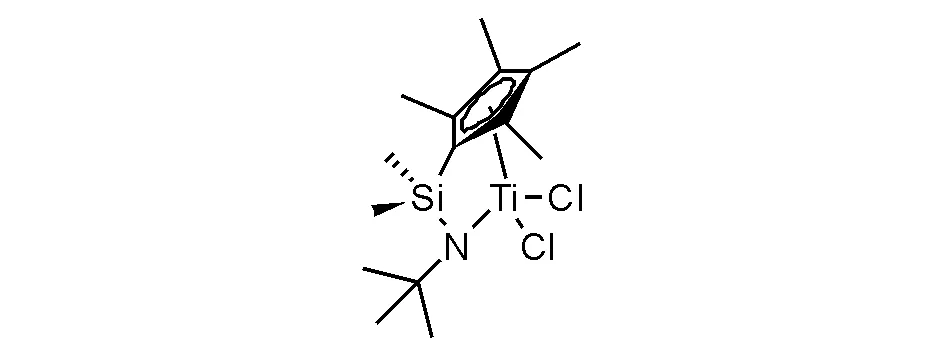
图 5-14 限定几何构型的催化剂
5.2.1.4后过渡金属配合物催化剂
20世纪90年代中叶,α-二亚胺的镍或钯配合物(图5-15,A)[35]和2,6-二亚胺吡啶的铁或钴配合物(图5-15,B)[36-37]能够实现乙烯聚合制备高分子量聚乙烯,由于后过渡金属配合物制备简单和稳定性高,很快吸引了学术界与产业界的广泛关注,并获得巨大的研究投入。图5-15中A和B两个模型是研究最多和获得衍生物最为丰富,基于相同的理论基础,发展了高效新型金属配合物模型(图5-15,C和D),呈现出更具实用价值的研究结果和新型聚合物材料。

图 5-15 典型后过渡金属催化剂模型
20世纪70年代,第三代Ziegler-Natta催化剂在催化活性上已经满足聚烯烃生产的需求,后续新型Ziegler-Natta催化剂和茂金属催化剂都是基于所得聚烯烃品质开展研究和用于改进生产。后过渡金属配合物催化烯烃聚合研究的突破口和意义,必然和应该集中在所得材料的性质与性能上。发现镍和钯配合物催化乙烯聚合过程中有更多氢转移的过程,获得了高度支化的聚乙烯甚至是聚乙烯弹性体[38];铁和钴配合物催化乙烯聚合获得高度线性的聚合物,而且在分子量低的聚乙烯中能够观察到清晰的端双键,成为制备不同高级α-烯烃的基石(作为新型长链共聚单体)和乙烯齐聚的高效催化剂[39]。相关的研究和产业化仍在进行中,所制备的新颖聚乙烯树脂有可能影响到新颖高性能树脂和材料的发展,帮助提高人们的生活水平和减少对环境的影响。
5.2.2合成橡胶材料及其催化聚合
在三大合成高分子材料中,橡胶占据了无可替代的地位,曾被作为军用和国防物资受到各国政府的重视,国际橡胶统计网会不断更新橡胶与弹性体的生产与应用。产量最大的三种合成橡胶依次分别是苯乙烯-丁二烯共聚物(styrene-butadiene rubber,SBR,简称为丁苯橡胶)年产三百多万吨、顺式-丁二烯橡胶(butadiene rubber, BR,顺丁橡胶)年产两百多万吨和异戊橡胶(isoprene rubber, IR)年产接近一百五十万吨,弥补了天然橡胶在产量和特殊性能需求上的不足。丁苯橡胶在1937年产业化,使用自由基聚合工艺,不在讨论之列。聚丁二烯橡胶和异戊橡胶具有相似的催化聚合过程和工艺,在此,我们使用比较成熟的丁二烯聚合催化剂进行讨论。
丁二烯聚合有两种方式和三种结构规整的聚合物(见图5-16),其中1,2-聚合所得聚合物含有大量的乙烯基,很容易交联,不过这类聚合少,所得聚合物敏感,难有应用价值。1,4-聚合所得聚丁二烯又基于其结构趋向性获得了反式聚丁二烯和顺式聚丁二烯,基于聚合反应自身推动力,获得顺式聚丁二烯(顺丁胶)结构聚合物更为普遍,其反式聚丁二烯认为是聚丁二烯产物中可以调节性能差异的组分,高顺式含量的聚丁二烯是产业界努力追求的。
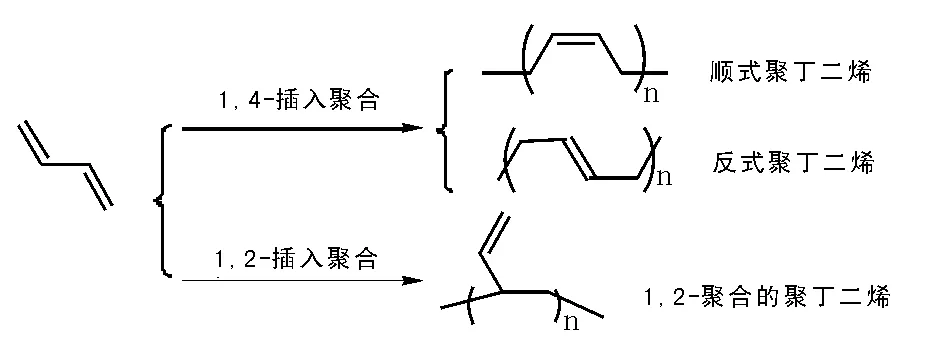
图 5-16 丁二烯的聚合与所得聚合物
顺丁橡胶是耐冲击的热塑性材料,具有优异的回弹性能和低的玻璃化温度(Tg为-110 ℃),耐寒和耐磨性能好。与天然橡胶相比,其耐热与抗老化性能更为优异,广泛应用于轮胎、传送带、软管卷涵盖、鞋底和高尔夫球等,不仅如此,还广泛用于其他热塑性材料的改性,添加到高抗冲聚苯乙烯和高抗冲丙烯腈-苯乙烯-丁二烯共聚物(ABS树脂)中,提高材料的性能,是重要的战略物资。聚丁二烯主要采用溶液聚合工艺,通常使用镍系、钴系和钕系催化剂;其中,镍系催化剂形成了成熟的催化体系,并获得广泛应用,钴系催化剂高顺式选择性特点备受瞩目,作为稀土大国更希望充分利用钕催化剂高活性和高选择性获得顺丁橡胶生成的新突破。
工业上镍系催化剂是三组分催化体系,主催化剂可以是乙酰丙酮镍、水杨酸镍、硬脂酸镍和环烷酸镍等;助催化剂由两部分组成,其一是以三异丁基铝、氢化二异丁基铝或甲基铝氧烷为主的烷基铝化合物,其二是如三氟化硼为主的氟化物。此外,通常镍系催化体系需要微量水参与陈化处理,镍与铝的比例一般为3~4,铝与丁二烯比为0.23~0.35,水与丁二烯则接近等物质的量,这类催化剂配方是常用的催化剂体系。不难看出,镍使用量较多,其催化活性提高是科学研究和技术发展的动力之一;文献中有很多主催化剂研究的例子,Ai P等[40]研究的吡啶胺醇配合物模型(图5-17)值得提及,其镍配合物能够室温下可以引发聚合得到顺式含量约为91%的聚丁二烯产品。

图 5-17 新型金属配合物丁二烯聚合催化剂
钴系催化剂是两组分催化体系,钴盐或者化合物为主催化剂,烷基铝为助催化剂,能够通过调整催化剂结构和控制聚合条件实现调控聚丁二烯产品的顺式含量和结构选择性,影响产物的宏观性能。例如,使用甲基铝氧烷助催化剂的体系中卤化钴和羧酸钴所得聚合物为高顺式聚丁二烯[41];然而,额外添加烷基膦或吡啶衍生物后,得到以1,2结构为主的聚丁二烯产物[42]。使用吡啶胺亚醇的钴配合物(图5-17),添加少量乙基倍半铝(EASC),能够对丁二烯实现顺式选择性的高效聚合;室温下,2000倍的丁二烯能在1 h内完全转化,聚合物的分子量为(2.31×105~3.08×105) g·mol-1,分子量分布为1.45~1.81,顺式-1,4含量为96.3%~96.9%[43]。
稀土氧化物最先用于乙烯聚合[44],随着研究深入,发现能够更为有效地应用于二烯聚合,因而,部分学者将二烯聚合催化剂称为Ziegler-Natta催化剂。 常用的钕催化体系由三组分构成,包括钕化合物主催化剂,还有烷基铝化合物助催化剂以及活化剂;常见的助催化剂有三甲基铝(TMA)、三乙基铝(TEA)、三异丁基铝(TIBA)、氢化二异丁基铝(DIBAH)、甲基铝氧烷(MAO)或者二丁基镁(MgBu2)等,活化剂主要是乙基倍半铝(EASC)、二氯化乙基铝(DEAC)、氯化二乙基铝、四氯化硅(SiCl4)和溴化铝(AlBr3)等。最初的钕催化丁二烯聚合容易产生凝胶,凝胶的存在影响聚合物性能。经过大量研究和筛选,目前具有实用价值的钕催化剂多是羧酸钕化合物(图5-18)[45]。
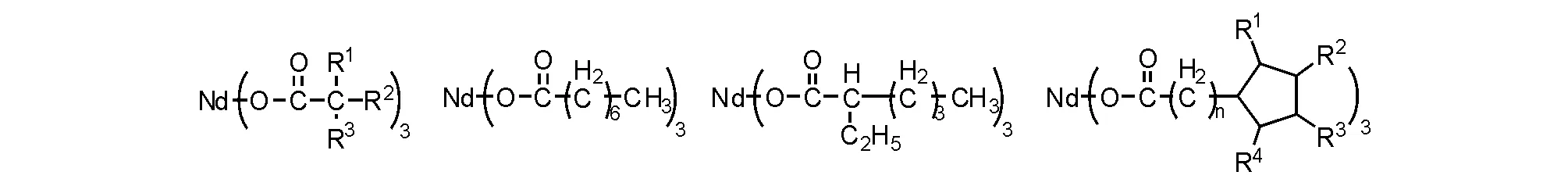
图 5-18 常用的羧酸钕催化剂结构
依据使用主催化剂金属的不同,又称为镍胶、钴胶和稀土(钕)胶。目前,工业级顺丁橡胶的主流产品还是镍胶和钴胶,其共同优点是催化剂活性高,产品中顺式-1,4结构含量高,质量均匀,由此制得橡胶制品的综合物理与机械性能较好。近十多年里快速发展的钕系催化剂获得产业化发展,稀土顺丁橡胶分子链拥有很高的规整度和线性结构,抗湿滑能力得到明显改善。
5.2.3合成聚酯材料及其催化聚合
聚酯是由多元醇和多元羧酸缩聚所得,是广泛应用的合成高分子材料,用于制备聚酯纤维和薄膜等。事实上,20世纪20年代DuPont公司和英国皇家化学工业公司(ICI)达成协议,共享特定合成材料的研究成果,因而,DuPont公司的研究成果很容易被英国学者获得详尽信息。基于DuPont公司的 Wallace Carothers博士在二酸与二胺反应制备具有里程碑意义的尼龙基础上,1941年英国化学家John Rex Whinfield和James Tennant Dickson最先申请对苯二甲酸和乙二醇缩聚(图5-19)制备聚对苯二甲酸乙二酯的专利,该催化技术于1946年在英国实现
产业化生产,1953年在世界范围大规模工业化生产。在过去半个多世纪,虽然使用了多种二酸和二醇进行缩聚反应获得高分子聚合物,其新材料的性能一直难于与已有的涤纶相媲美,因而,更多的研究与产业化集中在涤纶制备催化剂的研究;然而,抽象地考虑缩聚反应本身,就是缩合脱水过程和所得聚酯材料聚合度的控制问题。

图 5-19 苯二甲酸和乙二醇缩聚反应制备涤纶
目前,涤纶年产超过50 Mt,其中33 Mt用于制备聚酯纤维。涤纶合成乙二醇外,对苯二酸根的原料是对苯二甲酸或者对苯二甲酸二甲酯;对苯二甲酸二甲酯虽然在反应过程中会有甲醇副产物产生,但原料便于储运和提纯,是制备高品质涤纶所通用的。对于缩聚反应机理研究表明,该缩聚反应是通过酯化或脂交换反应,首先获得苯二甲酸缩二乙二醇中间体,该中间体进一步进行缩合脱去一个乙二醇,形成聚对苯二甲酸乙二酯(图5-20)。

图 5-20 涤纶合成机理
根据复合反应驱动力,只要能够加快第一步酯交换,或者酯化反应脱出的甲醇,或者水被转移与吸收,就比较容易地推动苯二甲酸缩二乙二醇中间体的形成;缩聚反应副产物是反应原料乙二醇,这步缩聚反应成了制备涤纶的控制步骤,其催化剂研究成为制备涤纶过程中研究的中心环节。涤纶生产所用的催化剂包括锑系、锗系和钛系三类,所得涤纶性能也略有差异。锗系催化剂所得涤纶呈现白色,具有切片性能优异和结晶速率快的优点,但存在氧稳定性差和热稳定性一般,锗的价格昂贵,除了满足光学材料和高透明材料需求外,难于大规模生产。钛系催化剂所得涤纶只是结晶速率好,其色泽和热氧稳定性都较差,但钛的低毒性质还是帮助所得涤纶材料一定的应用。尽管锑的残留可能导致白血病以及影响肝脏和脾脏的代谢,但是锑廉价和所得涤纶良好的结晶与热氧稳定性,还是使这类聚酯获得最为广泛的应用,占据了聚酯生产的八成以上。除此之外,还有铝系和锡系等催化剂体系在产业化研究和推进中。
乙二醇锑被认为是最为普遍应用的工业催化剂,其聚集态结构获得了单晶X-光晶体衍射的确认(图5-21)。其用于催化乙二醇与对苯二甲酸或者对苯二甲酸二甲酯进行缩聚制备涤纶过程中呈现锑离子外仅仅是乙二醇的存在,不会干扰反应物纯度,实现稳定生产的效果。此发现后,以往的锑系催化剂(三氧化二锑或者醋酸锑)逐渐被替代掉;与此同时,不难理解以往使用的锑催化剂很可能在体系中自动转化为乙二醇锑催化物种。

图 5-21 乙二醇锑的结构
为了开发替代锑系催化剂的新型高效和低(无)毒催化剂体系,近些年比较活跃的研究课题是钛系催化剂体系,主要还是使用钛酸脂(如钛酸丁酯或者钛酸异丙脂等)以及钛酸芳香脂和复合脂类化合物,力求提高催化聚合活性和降低钛在聚酯中的残留,改善聚酯的色度;不仅如此,还希望提高所得聚酯的分子量和降低分子量分布,以便提高聚酯的耐氧和耐热稳定性。总之,聚酯生产了70年,其需求和生产的发展仍然持续,并期望在低毒聚酯生产技术上获得突破。
5.3展望
高分子合成发展近一个世纪,使人们的居住、交通设施乃至食品包装和保鲜都发生了极大的变革,使得人类社会逐渐摆脱了依赖天然材料和无机材料的历史。在经历探索和发展阶段后,高分子材料的广泛应用和普遍推广满足了对于“自然资源消耗减量,但社会繁荣和人们生活高水准”的要求,替代自然材料不仅保护了自然资源而且减少了防腐和成品变型的缺点,满足了人们对于审美的要求,提高了成品的质量。随着化学和物理学的认知发展,新材料研究和持续发展仍在进行中,其合成和制备工艺中减少副产物产生和高分子材料微结构控制都可以借助催化效率提高和高效催化剂的研制实现。在量大面广的高分子合成中,任何合成与加工工序中微小的效率提高都将极大地提升产业的附加值。本章节列举的只是三个大品种传统高分子材料,并没有介绍特种高分子材料和高分子专用料材料的催化问题。就文中涉及的聚烯烃催化问题,被称为“塑料之王”的超高分子量聚烯烃材料其在北京生产所用催化剂仅是在普遍使用的Ziegler-Natta催化剂基础上条件控制实现的,这种条件控制却丧失了产量效益,只是常规聚烯烃生产量的三分之一;尽管目前超高分子量聚乙烯每年只有数十万吨的需求,一旦新型高效催化剂出现降低了生产成本,在合理价格基础上肯定会有飞跃性应用发展。常规聚烯烃生产的催化剂虽然成熟,聚烯烃新材料特别是后过渡金属催化剂制备的聚乙烯新材料还刚刚起步;无论高度线性聚乙烯材料还是高度支化聚乙烯材料都是以往产业界所不能预测的新材料,高度支化聚乙烯材料一旦可以添加部分二烯共聚单体,就可能引发橡胶新材料的革命。目前二烯催化合成橡胶的催化效率还普遍比较低,催化剂在橡胶及其制品中的残留值都很高,硫化交联后仍有很大部分双键成为橡胶制品老化的推进剂;橡胶合成的高效催化剂具有迫切需要。尽管锑的毒副作用获得普遍关注,锑系聚酯中每克聚酯的锑残留仍为(120~350) μg,而且这类聚酯呈现浅灰色;这种色泽的缺点与快速发展的聚酯市场形成巨大的差异,更难于满足人们对于降低金属(毒副作用)残留的需求,必将需要一场聚酯催化剂的新革命。
不仅如此,合成高分子材料使用后的有效利用和污染控制问题形成了另一个方兴未艾的催化课题:高分子材料降解成为有机小分子,甚至是高分子合成的单体;这将有利于形成合成高分子材料“制备-使用-处理”的良性循环。合成,阳光下的上帝之路;降解,夜幕中的物能之智。师从自然,人为加速;自然界植物不断进行光合作用提供材料,人类与动物不断消耗和分解,形成完美循环。催化角度看问题,数千年前人们就知道了“酿酒”和“发酵”的菌种这类生物催化剂。目前,出现了高分子材料生物降解和催化降解两个有效途径,在“时间就是效率”的背景下,需要筛选出高效和快速的降解途径,化学工程的热裂解和催化降解除了仍需不断找寻高效催化剂外,制约技术的瓶颈还是高分子材料的回收和分类集中的问题。
总之,合成高分子材料以及其催化剂与工艺的研究发展是目前和未来半个世纪材料科学革命的基础,而材料革新必将促进交通运输工具和工程技术的飞跃发展。无论从专业角度还是社会需求,都为合成高分子材料相关的催化问题研究提供了广阔的空间,并为社会和每个参与者提供了光明的未来。
参考文献:
[1]Goodyear C.Improvement in India-rubber fabrics:US,3633[P].1844-06-15.
[2]BrydsonJ A.Plastics materials[M].6th Edition,Oxford:Butterworth-Heinemann,1995.
[3]Baekeland L H.Chemical achievers:the human face of chemical sciences,chemical heritage foundation(2005)[M].Retrieved.2007-11-08.
[4]Fawcett E W.The formation of high polymers[J].Journal of the Chemical Society,1936,32:119-121.
[5]Hogan J P,Banks R L.Polymerization of olefins:US,2825721[P].1954-08-21.
[6]Clark A.Polymerization and copolymerization of liefins on chromium oxide catalysts[L].Advances in Chemical Physics,1969,91:387-397.
[7]Ruddick V J,Badyal J P S.CO reduction of calcined CrOx/SiO2ethene polymerization catalysts[J].Langmuir,1997,13:469-472.
[8]McDaniel M P,Collins K S,Benham E A,et al.The activation of Phillips Cr/silica catalysts Ⅴ.Stability of Cr(Ⅵ)[J].Applied Catalysis A:General,2008,335:252-261.
[9]Cossee P.Ziegler-Natta catalysisⅠ.Mechanism of polymerization of alpha-olefins with Ziegler-Natta catalysts[J].Journal of Catalysis,1964,3:80-88.
[10]Gröneveld C,Wittgen P P M M,Swinnen H P M,et al.Hydrogenation of olefins and polymerization of ethylene over chromium-oxide silica catalystsⅤ.In situ infrared measurements and investigation of the polymer[J].Journal of Catalysis,1983,83:346-361.
[11]Ellermann V J,Hagen K,Krauss H L.Chemistry of polyfunctional ligands:complexes of chromium(Ⅱ)-chloride and surface chromium(Ⅱ) with polydentatep-ligands and as-ligands[J].Zeitschrift Fur Anorganische Und Allgemeine Chemie,1982,487:130-140.
[12]McDaniel M P.Supported chromium catalysts for ethylene polymerization[J].Advances in Catalysis,1985,33:47-98.
[13]Ziegler K,Holzkamp E,Martin H.Polymerisation vonthylen und anderen olefinen[J].Angewandte Chemie,1955,67:426-426.
[14]Ziegler K,Holzkamp E,Martin H.Das mülheimer normaldruck-polyäthylen-verfahren[J].Angewandte Chemie,1955,67:541-547.
[15]Natta G,Pino P,Corradini P,et al.Crystalline high polymers of alpha-olefins[J].Journal of the American Chemical Society,1955,77:1708-1710.
[16]Boor J.Ziegler-Natta catalyst and polymerization[M].New York:Academic Press,1979.
[17]Ivin K J,Rooney J J,Stewart C D,et al.Mechanism for stereospecific polymerization of olefins by Ziegler-Natta catalysts[J].Journal of the Chemical Society,1978,14:604-606.
[18]Green M L H.Studies on synthesis,mechanism reactivity of some organo-molybdenum and organo-tungsten compounds[J].Pure and Applied Chemistry,1978,50:27-35.
[19]Piers W E,Bercaw J E.Alpha-agostic assistance in Ziegler-Natta polymerization of olefins-deuterium isotopic perturbation of stereochemistry indicating coordination of an alpha-C—H bond in chain propagation[J].Journal of the American Chemical Society,1990,112:9406-9407.
[20]Galli P,Barbe P C,Noristi L.High-yield catalysts in olefin polymeriztion-general outlook on theoretical aspects and industrial uses[J].Angewandte Makromolekulare Chemie,1984,120:73-90.
[21]Galli P,Luciani L,Cecchin G.Advances in the polymerization of polyolefins with coordination catalysts[J].Angewandte Makromolekulare Chemie,1981,94:63-89.
[22]Galli P,Milani F,Simonazzi T.New trends in the field of propylene based polymers[J].Polymer Journal,1985,17:37-55.
[23]Busico V.Giulio Natta and development of stereoselective propene polymerization[J].Advances in Chemical Physics,2013,257:37-57.
[24]Natta G,Pino P,Mazzanti G,et al.Polimerizzazione stereospecifica delle alfa-olefine Nota[J].Gazzetta chimica Italiana,1957,87:549.
[25]Breslow D S,Newburg N R.Bis-(cyclopentadienyl)-titanium dichloride-alkylaluminum complexes as catalysts for the polymerization of ethylene[J].Journal of the American Chemical Society,1957,79:5072-5073.
[26]Chien J C W.Kinetics of ethylene polymerization catalyzed by bis-(cyclopentadienyl)-titanium dichloride dimethylaluminum chloride[J].Journal of the American Chemical Society,1959,81:86-92.
[27]Waters J A,Mortimer G A.Study of (pi-C5H5)2Ti(C2H5)Cl and its higher homologs in soluble Ziegler catalysts[J].Journal of Applied Polymer Science,1972,10:895-907.
[28]Sinn H,Patat F.Uber die wirkungsweise metallorganischer katalysatoren[J].Angewandte Chemie,1963,75:805-813.
[29]Sinn H,Bandermann F,Hinck H A.Polymerization reaction of trimethylaluminum [J].Angewandte Chemie International Edition,1968,7:399-414.
[30]Kaminsky W,Kopf J,Sinn H,et al.Extreme bond angle distortion in organozirconium compounds active toward ethylene[J].Angewandte Chemie International Edition,1976,15:629-630.
[31]Heins E,Hinck H,Kaminsky W,et al.Examples of condensation reactions of emthyl alkyls with cleavage of alkanes[J].Makromol Chem,1970,134:125.
[32]Andresen A,Cordes H G,Herwig J,et al.Halogen-free soluble Ziegler catalysts for polymerization of ethylene-control of molecular-weight by choice of temperaure[J].Angewandte Chemie International Edition,1976,15:630-632.
[33]Glaser R,Sun X.Thermochemistry of the initial steps of methylaluminoxane formation.Sluminoxanes and cycloaluminoxanes by methane elimination from dimethylalumium hydroxide and its dimeric aggregates[J].Journal of the American Chemical Society,2011,133:13323-13336.
[34]Kaminsky W.New polymers by metallocene catalysis[J].Macromolecular Chemistry and Physics,1996,197:3907-3945.
[35]Johnson L K,Killian C M,Brookhart M.New Pd(Ⅱ)-based and Ni(Ⅱ)-based catalysts for polymerization of ethylene and alpha-olefins[J].Journal of the American Chemical Society,1995,117:6414 -6415.
[36]Small B L,Brookhart M.Highly active iron and cobalt catalysts for the polymerization of ethylene[J].Journal of the American Chemical Society,1998,120:4049-4050.
[37]Britovsek G J P,Gibson V C,Kimberley B S,et al.Novel olefin polymerization catalysts based on iron and cobalt[J].Chemical Communications,1998,63:849-850.
[38]Wang S,Sun W H,Redshaw C.Recent progress on nickel-based systems for ethylene oligo-/polymerization catalysis[J].Journal of Organometallic Chemistry,2014,751:717-741.
[39]Ma J,Feng C,Wang S,et al.Bi- and tri-dentate imino-based iron and cobalt pri-catalysts for ethylene oligo-/polymerization[J].Inorganic Chemistry Frontier,2014,1:14-34.
[40]Ai P,Chen L,Guo Y,et al.Polymerization of 1,3-butadiene catalyzed by cobalt(Ⅱ) and nickel(Ⅱ) complexes bearing imino- or amino-pyridyl alcohol ligands in combination with ethylaluminum sesquichloride[J].Journal of Organometallic Chemistry,2012,705:51-58.
[41]Nath D C D,Shiono T,Ikeda T.Cis-specific living polymerization of 1,3-butadiene with CoCl2and methylaluminoxane[J].Macromolecular Chemistry and Physics,2002,203:756-760.
[42]Ricci G,Sommazzi A,Masi F,et al.Well-defined transition metal complexes with phosphorus and nitrogen ligands for 1,3-dienes polymerization[J].Coordination Chemistry Reviews,2010,254:661-676.
[43]Jie S,Ai P,Li B G.Highly active and stereospecific polymerization of 1,3-butadiene catalyzed by dinuclear cobalt(Ⅱ) complexes bearing 3-aryliminomethyl-2-hydroxybenzaldegydes[J].Dalton Trans,2011,40:10975-10982.
[44]Brown C P,Saunders J.Polymerization Catalysts for ethyleneⅠ.Rare earth oxides [J].Journal of Polymer Science,1960,43(142):579-579.
[45]Friebe L,Nuyken O,Obrecht W.Neodymium-based Ziegler/Natta catalysts and their application in diene polymerization[J].Advances in Polymer Science,2006,204:1-154.
孙文华,1963年生,博士,无党派民主人士,中国科学院化学研究所二级研究员和中国科学院大学岗位教授。
1986年在兰州大学完成本科学习后,1986-1989年在中国科学院兰州化学物理研究所完成硕士学位并留所工作,1991-1994年在中国科学院兰州化学物理研究所完成在职博士学习;同期,1993年12月经中国科学院批准在中国科学院兰州化学物理研究所任副研究员。1995年11月-1999年10月在日本学术振兴会、日本科学技术事业团和日本文部省外国人客座教授资助下在日本北海道大学催化中心工作;1999年10月在中国科学院“百人计划”支持下到中国科学院化学研究所任研究员。其兰州工作集中在羰基原子簇设计合成和氢甲酰化催化,日本工作集中在有机合成方法学。此外,作为洪堡基金会访问教授在德国明斯特大学、日本文部省访问教授在名古屋大学、以及法国路易斯-帕斯卡大学和斯特拉斯堡大学访问教授,进行访问研究。在中国科学院化学研究所的工作集中开展“过渡金属配合物烯烃聚合”催化剂与聚合工艺,以及新型聚烯烃材料研究;发表研究论文300余篇,获得授权专利57件,是爱思唯尔统计2014年和2015年“中国高被引化学研究者”之一。基于其研究成果,获得了“2009年度北京市科技进步一等奖”,“2011年中国石油和化学工业联合会科技进步奖一等奖”,2011年当选为英国皇家化学会会士,2012年获得中国侨界创新人才贡献奖,2013年获得第七届冯新德高分子奖最佳文章奖。
现代催化化学讲座
