纳米储氢材料在推进剂中的应用研究进展
2016-05-09杨燕京赵凤起仪建华
杨燕京, 赵凤起, 仪建华, 罗 阳
(西安近代化学研究所燃烧与爆炸技术重点实验室, 陕西 西安 710065)
1 引 言
近年来,储氢材料在推进剂中的应用得到了学术界的广泛关注[1- 2]。固体推进剂中的储氢材料在推进剂燃烧时将受热分解并放出氢气(H2)。作为分子质量最小的气体,H2降低了燃烧室内燃气的平均分子质量,从而提高了推进剂的比冲; 另一方面,H2通过燃烧释放出大量的能量,进一步改善了推进剂的能量水平。储氢材料对固体推进剂能量性能的影响与其放氢量和放氢行为密切相关。其中,储氢材料的理论放氢量由其成分决定,而实际放氢量则受到放氢性能的影响。因此,放氢性能在很大程度上决定了储氢材料调节推进剂能量水平的效果。储氢材料的放氢性能由放氢热力学性能和放氢动力学性能两个指标表征。为了充分利用其中所储存的氢,储氢材料的热稳定性应与推进剂的燃烧温度分布、推进剂组分的热稳定性和推进剂加工工艺等相匹配,即其应具备合适的放氢热力学性能。此外,固体推进剂的燃烧是一个反应速率较快的氧化还原过程,这要求其中的储氢材料能以较快的速率放出H2,即储氢材料应具有优异的放氢动力学性能。在固体推进剂中的应用对储氢材料的放氢热力学和动力学性能提出了较高的要求; 若储氢材料的放氢热力学和动力学性能不满足要求,则其储存的氢无法在推进剂燃烧时及时转化为氢气,而将以其它方式参与推进剂的燃烧; 这与将储氢材料引入推进剂的初衷相悖,不利于推进剂比冲的提高。
按照储氢材料与氢作用机理的不同,储氢材料可以分为化学吸附储氢材料、物理吸附储氢材料以及化学-物理吸附储氢材料[3]。其中,轻金属氢化物、配位氢化物和化学氢化物等化学吸附储氢材料储氢量较高[4],在推进剂领域有较大的应用潜力; 然而,此类材料的放氢热力学和动力学性能一般较差,不能满足在推进剂中的应用要求。纳米化可以缩短化学吸附储氢材料放氢反应的传质路径,改善动力学性能; 同时,纳米化可以使得材料的比表面积显著增大,导致材料的表面能增大而形核活化能减小,从而降低化学吸附储氢材料放氢反应的能垒。因此,纳米化可以在不引入其它物质的前提下调节化学吸附储氢材料的本征性质,从而显著改善材料的放氢热力学和动力学性能,适用于改善推进剂中储氢材料的放氢性能。对于以物理吸附方式储氢的多孔碳、沸石、金属有机框架化合物(MOFs)、共价有机框架化合物(COFs)以及自具微孔聚合物(PIM)等材料来说,它们本身就具备纳米结构特征,具有很好的储氢动力学性能。然而,物理吸附储氢材料的储氢热力学性能较差,即它们吸放氢过程的焓变过小,难以在室温下稳定储氢。对于此类材料,通过调节其纳米结构特征以及在纳米尺度上对材料的孔道进行修饰等手段,可以提高其吸放氢焓变,实现较高温度下的储氢。
本文综述了不同种类纳米储氢材料的研究现状、发展方向,并展望了其在固体推进剂中的应用前景。
2 化学吸附储氢材料
化学吸附储氢材料的纳米化可以通过三类方法实现,第一类方法利用机械力将普通储氢材料粉碎至纳米尺度,例如各种机械球磨方法; 第二类方法是直接合成纳米级的储氢材料; 第三类方法则是使用具有纳米孔道的载体,将储氢材料负载于其中,从而实现化学吸附储氢材料的纳米化。本节将从化学吸附储氢材料纳米化方法的分类出发,论述纳米化学吸附储氢材料的研究现状。
2.1 机械力粉碎
在储氢材料研究中,以机械球磨为代表的机械力粉碎是最常见的纳米化方法之一,其原理是通过材料与磨球或是材料与材料之间的撞击、摩擦等相互作用减小材料的晶粒尺寸和颗粒尺寸,属于物理方法。
对于不同种类的化学吸附储氢材料来说,机械球磨均能有效降低其晶粒尺寸和颗粒尺寸,特别是可以将材料的晶粒尺寸降低至数纳米。然而,需要指出的是,若不引入助剂或分散剂,使用机械球磨法难以得到颗粒尺寸较小的样品。这是因为当颗粒尺寸降低到一定程度时,其表面能很高,在球磨条件下很容易发生颗粒的长大和团聚。尽管如此,球磨法仍然是一种降低储氢材料放氢温度、提高其放氢动力学性能的有效方法。
研究人员发现,与普通氢化镁相比,经球磨处理的MgH2拥有更好的吸放氢性能,其原因在于球磨减小了氢化镁的颗粒尺寸。Markmaitree等[5]利用机械球磨对氨基锂(LiNH2)进行处理,将其晶粒尺寸降低至5.5 nm,从而相应地将LiNH2分解的起始温度由120 ℃降低至室温,同时LiNH2分解的活化能也由243.98 kJ·mol-1降低到138.05 kJ·mol-1。Liu等[6]系统研究了颗粒尺寸对于Li2MgN2H2储氢材料性能的影响,发现颗粒尺寸的减小能显著改善吸放氢的动力学性能。普通Li2MgN2H2的放氢反应活化能为132.6 kJ·mol-1,而颗粒尺寸为100~200 nm的Li2MgN2H2的活化能为51.6 kJ·mol-1,降低了约61%。
引入球磨助剂可以避免球磨过程中颗粒的团聚和长大,从而获得颗粒尺寸较小的储氢材料。Paskevicius等[7]在高能球磨过程中引入大量LiCl基质,将MgH2的颗粒尺寸降至7 nm。颗粒尺寸的减小使得MgH2的分解焓由74.06 kJ·mol-1降低至71.22 kJ·mol-1,显著改善了MgH2的放氢性能。尽管球磨助剂能促进储氢材料的纳米化,但大量助剂的存在会导致储氢材料纯度的降低和储氢量的下降; 若增加提纯步骤以除去助剂,就会提高工艺的复杂度。
作为一种成熟的储氢材料纳米化技术,机械球磨能较容易地实现低成本、大批量的纳米储氢材料制备,从储氢材料在推进剂中应用的角度来说具有明显优势。但是,该方法难以获得颗粒尺寸较小以及具备特殊微观形貌的储氢材料,在一定程度上限制了其应用。
2.2 纳米储氢材料的直接合成
与机械粉碎“由上到下”的纳米化思路不同,纳米储氢材料的直接合成属于“由下到上”的纳米化方法,通过控制物质的形成过程,直接获得纳米级别的储氢材料。
气相沉积是一种常见的纳米材料和纳米结构的制备方法。在储氢材料研究领域,研究人员在利用气相沉积制备MgH2纳米线等纳米结构方面已做了较多工作,该方法在制备复杂氢化物中也得到了成功应用。Li等[8]使用气相沉积制备了直径为30~50 nm、80~100 nm和150~170 nm的Mg纳米线,随后将其氢化以获得MgH2纳米线。性能测试发现,上述MgH2纳米线的放氢动力学性能随着直径的减小而加强,直径为30~50 nm、80~100 nm和150~170 nm的MgH2纳米线的放氢反应活化能分别为38.8,46.5 kJ·mol-1和81.1 kJ·mol-1,均表现出了优异的吸放氢性能。基于密度泛函理论的计算结果证实,MgH2纳米线尺寸的减小起到了去稳定化效果,这是MgH2性能获得改善的主要原因。Zhu和Saita等[9]使用类似的方法制备了MgH2纳米纤维,并发现氢气气氛的压力对所得产物的形貌有明显影响。在进一步的研究中,Zhu等[10]系统研究了温度、氢气压力和沉积过程等对气相沉积合成MgH2微米或纳米线的影响进行了系统研究,并由此给出了合成MgH2纳米线的最优条件。Pang等[11]提出了一种基于物理气相沉积方法的铝氢化镁(Mg(AlH4)2)纳米线的合成方法,该方法的特殊之处在于通过球磨Mg(AlH4)2的乙醚加合物Mg(AlH4)2·2Et2O而产生蒸汽,蒸汽在球磨罐内的特殊装置处沉积而得到Mg(AlH4)2纳米线。性能表征结果证实,Mg(AlH4)2纳米线的分解放氢温度比普通Mg(AlH4)2有所降低,放氢动力学性能也获得了改善。
以惰性纳米材料作为模板,可以合成具备相同形貌的纳米储氢材料。该方法的优点在于,调节模板的尺寸,即可方便地调节所得纳米储氢材料的尺寸。NaAlH4的含氢量可达7.5%,但其放氢温度偏高,放氢动力学性能较差。Balde等[12]以碳纤维为模板,合成了系列纳米铝氢化钠(NaAlH4)储氢材料。对使用模板制备的纳米NaAlH4储氢材料的性能表征显示,材料的放氢温度随着材料粒径的减小而降低,这一发现进一步证明了纳米化对于改善储氢材料放氢性能的作用。
Xie等[13]采用等离子体电弧合成了亚氨基锂(Li2NH)的空心纳米球。他们在Ar/NH3混合气中用电弧蒸发金属锂(Li),即可获得LI2NH的纳米球。所得Li2NH纳米球的直径在100~400 nm范围内(90%以上的球直径在100~200 nm范围内),球壁厚约20 nm。与普通的Li2NH相比,该Li2NH纳米球的吸放氢反应温度都有所降低,吸放氢动力学性能都获得了提高,其中Li2NH纳米球的放氢温度与普通样品相比降低了120 ℃。随后,Xie等[14]使用类似的方法制备了氨基镁(Mg(NH2)2)的纳米球。他们首先使用等离子体电弧法制备了Mg3N2纳米颗粒,随后使之与NH3反应,从而获得Mg(NH2)2纳米球。对Mg(NH2)2纳米球与氢化锂(LiH)组成的储氢体系性能的表征发现,其放氢温度随着纳米球直径的减小而显著降低[15]。此外,研究人员测得含直径为2000,500,100 nm的Mg(NH2)2纳米球的体系的放氢活化能分别为182.0,134.7,122.2 kJ·mol-1[15],说明其放氢动力学性能随着颗粒尺寸的减小也获得了提高。
静电纺丝是近年来受到广泛关注的一种纳米纤维材料制备方法,Kurban等[16]使用共轴静电纺丝制备了氨硼烷(NH3BH3)-聚苯乙烯的纤维(图1),并研究了其放氢性能。通过改变所使用的溶剂和实验条件,可以获得不同形貌和结构的纤维。与普通的NH3BH3相比,氨硼烷-聚苯乙烯纤维的分解放氢温度降低了15~20 ℃[16],放氢性能获得了改善。
纳米储氢材料的直接合成可以通过多种方法实现,所得到的材料在形貌、结构和性能等方面也呈现出丰富的多样性。此类方法适用于“按需合成”,即根据对性能的要求针对性地制备具有一定形貌和结构特征的材料。然而,与成熟的机械力粉碎方法相比,此类方法的成本较高,同时难以实现材料的规模化制备,这都限制了此类方法的实际应用。

图1氨硼烷-聚苯乙烯纤维扫描电镜图[16]
Fig.1SEM images of the NH3BH3-polystyrene nanofibers[16]
2.3 纳米限域
纳米限域也是一种常见的储氢材料纳米化的方法,该方法利用载体内部纳米尺度的孔道来承载材料,通过孔道内壁的限制而实现材料的纳米化。通过选择成分、孔径和孔道特征不同的载体,可以灵活地调控所承载的储氢材料的吸放氢性能。纳米限域储氢材料的制备主要利用溶剂将储氢材料或其前驱体溶解,或是在一定条件下加热使储氢材料或其前驱体融化,随后将溶液或是熔融物质渗入限域载体,再通过一定的后处理手段,即可得到目标产物。
与其它纳米化方法相比,使用纳米限域容易获得颗粒尺寸很小的储氢材料,同时还可以避免纳米储氢材料的团聚。因此,纳米限域已成为一种重要的储氢材料纳米化方法。
研究发现,将熔融的金属镁(Mg)或是二丁基锂渗入多孔碳的纳米级孔道中,随后使其与氢气(H2)反应,即可获得纳米限域的MgH2储氢材料[17]。与普通MgH2相比,纳米限域MgH2的放氢动力学性能获得了显著改善。
研究人员利用纳米限域手段,在纳米化改性NaAlH4放氢性能方面做了大量工作[18~21]。Baldé等[18]使用纳米纤维之间的空隙实现了对NaAlH4的纳米限域,所得NaAlH4的颗粒尺寸分别在1~10 μm、19~30 nm和2~10 nm范围内。性能测试发现,粒径为1~10 μm的NaAlH4的放氢温度为186 ℃,放氢活化能为116 kJ·mol-1; 而粒径为2~10 nm的NaAlH4的放氢温度为70 ℃,活化能为58 kJ·mol-1。可见,通过纳米限域实现纳米化可以显著改善NaAlH4的放氢性能,特别是放氢动力学性能。Fan等[19]使用孔径分别为200,60,30,4 nm的多孔碳作为载体,系统研究了载体孔径对于NaAlH4放氢性能的影响。研究发现,随着多孔碳孔径的下降,限域于其中的NaAlH4的融化温度和分解放氢温度都明显降低。孔径为200 nm的多孔碳中的NaAlH4放氢峰值温度为208 ℃,而孔径为4 nm的碳载体中的储氢材料在153 ℃即达到放氢峰值。此外,当多孔碳的孔径由200 nm降低至4 nm时,限域于其中的NaAlH4的放氢活化能也从124.3 kJ·mol-1降低至69.7 kJ·mol-1。此项工作证明,纳米限域所使用载体的孔径越小,对于储氢材料放氢性能的强化效果越好。此外,研究人员发现,除多孔碳外,金属有机框架化合物(Metal-organic Frameworks, MOFs)也是储氢材料纳米限域的优良载体。Bhakta等[22]选用MOF HKUST-1(Cu3(BTC)2)作为载体,研究了NaAlH4-HKUST-1储氢体系的性能。限域于HKUST-1中的纳米NaAlH4的放氢温度比普通NaAlH4降低了100 ℃,动力学性能也获得了一定改善。
除金属氢化物和金属铝氢化合物以外,金属硼氢化合物也是一类重要的高容量储氢材料。LiBH4的氢含量高达18.5%,是储氢量最高的金属硼氢化物。但是,LiBH4的稳定性较高,主要放氢反应需在400 ℃以上进行。已有研究成果证实,利用纳米限域手段将LiBH4纳米化,可以有效改善其放氢性能。Brun等[23]使用甲基叔丁基醚作为溶剂,通过湿法浸渍实现了LiBH4在多孔碳中的纳米限域。该工作中所使用的多孔碳的微孔孔径约为0.5 nm,限域于微孔中的LiBH4的分解放氢温度下降至约200 ℃,将其加热至300 ℃可释放出4%的氢,放氢性能获得了显著改善。使用孔径为2~26 nm的介孔碳气凝胶作为载体,对LiBH4进行纳米限域处理后,LiBH4的放氢热力学性能和动力学性能都获得了显著改善; 此外,改善的效果随着孔径的减小而增强[24- 25]。Vajeeston等[26]通过理论计算考察了LiBH4纳米簇和纳米晶须的稳定性。研究发现,LiBH4纳米簇和纳米晶须的尺寸若小于1.75 nm和1.5 nm,其稳定性会大大降低。此外,由于LiBH4纳米结构中大部分的原子都暴露于表面,而表面原子之间的作用力较弱; 因此,与普通LiBH4相比,纳米LiBH4更容易分解放氢。除LiBH4外,纳米限域技术在其他金属硼氢化物储氢材料中亦有应用。Fichtner[27]以孔径小于2 nm的活性炭作为限域载体,研究了纳米化的Mg(BH4)2的放氢行为。研究发现,纳米限域使得Mg(BH4)2的放氢峰值温度降低。此外,纳米限域后,Mg(BH4)2第一步放氢反应的活化能由310.7 kJ·mol-1降低至176.2 kJ·mol-1,显示出纳米化对其放氢动力学性能良好的改善作用。最近,Yan等人[28]通过纳米限域的MgH2-碳气凝胶材料与B2H6/H2混合气体的反应,制备了以碳气凝胶为载体的纳米限域Mg(BH4)2储氢材料。该纳米Mg(BH4)2的分解放氢温度显著降低,主要放氢过程在160 ℃即已开始。更重要的是,使用碳气凝胶进行纳米限域处理后,Mg(BH4)2的放氢活化能从340 kJ·mol-1降低至102 kJ·mol-1,仅为原来的1/3。
纳米限域技术在NH3BH3的储氢性能改进中也得到了应用。Gutowska等[29]使用介孔二氧化硅SBA-15作为限域载体,甲醇作为溶剂,将NH3BH3限域于孔径为7~8 nm的纳米孔中。纳米化后的NH3BH3的放氢温度有所降低,放出的氢气的纯度明显提高,放氢活化能也由184 kJ·mol-1降低至67 kJ·mol-1,显示出纳米化对于NH3BH3放氢性能的明显改善作用。Feaver等[30]则使用碳气凝胶作为NH3BH3纳米限域的载体,孔径为2~20 nm。使用碳气凝胶进行纳米限域后,NH3BH3的放氢反应由110 ℃和150 ℃处的两个放氢峰转变为90 ℃处的一个放氢峰,放氢热力学和动力学性能都获得了改善。
纳米限域技术在储氢材料放氢热力学和动力学性能的改善中扮演了重要的角色。但是,该方法的不足之处在于: 对储氢材料的纳米限域处理需要引入大量的多孔载体材料,会导致材料储氢量的显著降低。此领域的研究工作应着重于发展新型载体材料,在改善材料放氢热力学和动力学性能的同时尽量避免材料储氢量的降低。
3 化学-物理吸附储氢材料
化学-物理吸附储氢材料主要指一类纳米结构的无机材料,包括BN、MoS2、TiO2和ZnO2等。它们与氢的相互作用不仅受到材料化学性质的影响,也与其纳米结构密切相关,介于传统的化学吸附储氢材料和物理吸附储氢材料之间。
Wang等[31]在对氮化硼(BN)的研究中发现,在氢气气氛中球磨六方相氮化硼(h-BN) 80 h后,所得BN微粉的晶粒尺寸为3 nm,且含有2.6%的氢,即球磨使得BN纳米晶吸氢。Ma等人[32]则研究了不同结构BN纳米管(图2)的储氢性能,发现竹结构的BN纳米管的储氢量可达2.6%,远高于普通BN的0.2%; 此外,BN纳米管所储存的氢气在300 ℃可以完全放出。Tang等[33]则发现,塌陷的BN纳米管具有更高的储氢量,可达4.2%,这是因为其中有更多易与氢反应的缺陷。Oku和Kuno等人[34-35]则制备了一系列BN的纳米胶囊、纳米管和纳米笼,纳米胶囊和纳米管的混合物储氢量最高,可达3.2%。上述结果表明,纳米结构的BN具有不错的储氢能力和较低的放氢温度,但其储氢特性与微观结构密切相关,需通过调控合成方法以获得最优的储氢性能。
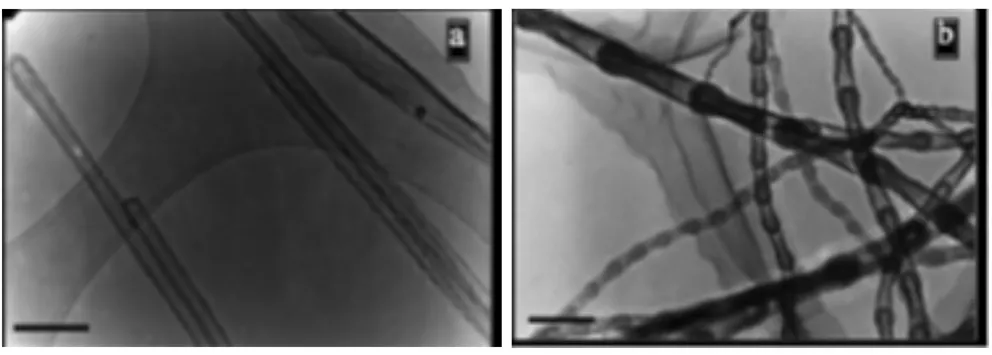
a.multiwall BN nanotubesb.bamboo-like BN nanotubes
图2不同微观结构的BN纳米管扫描电镜图片[32]
Fig.2SEM images of BN nanotubes with different,morphologies[32]
除纳米结构的BN外,研究人员发现某些纳米结构的金属氧化物和金属硫化物也可用作储氢材料。Wan等[36]发现,氧化锌(ZnO)纳米线可以储存0.83%的氢。在随后的研究中,Pan等[37]发现ZnO纳米线的储氢量可达2.57%; 若用Mg掺杂ZnO,可进一步将储氢量提高到2.75%。最近,基于密度泛函理论对TiO2纳米管进行的研究发现,单壁TiO2纳米管与H2的结合能为0.053 eV/H2分子,对应的储氢量可达3.2%[38]。除氧化物外,TiS2和MoS2等金属硫化物也具有一定储氢能力,其中TiS2纳米管可以储存2.5%的H2[38],而MoS2纳米管则可以储存1.2%的H2。
上述研究工作证实,通过一定的纳米化手段,可以获得包括纳米金属氧化物等具备储氢能力的纳米结构无机材料,这一发现在固体推进剂的研究中具有一定价值。铅(Pb)、铜(Cu)、铋(Bi)、铁(Fe)等的纳米氧化物是固体推进剂燃烧的高效催化剂,但它们是不含能的; 若能通过一定的合成方法调控等手段,使得铅(Pb)、铜(Cu)、铋(Bi)、铁(Fe)等的纳米氧化物具备储氢能力,可以使其吸附氢气而将其含能化,这有利于它们在固体推进剂中的应用。
4 物理吸附储氢材料
物理吸附储氢材料中,氢与固体材料间的作用力为范德华力(van der Waals forces); 只要温度高于氢的沸点(20.4 K),物理吸附即可发生。在势能曲线上,氢与固体材料物理吸附的势能最小值约在氢的分子半径处,即氢以单层形式吸附于固体表面。氢物理吸附的焓变多在4~8 kJ·mol-1,这意味着在低温下才可能获得可观的储氢量。通过控制材料的纳米微观结构,研究人员已经制备出具有较高储氢量的物理吸附储氢材料。在77 K和2×106Pa下,使用聚吡咯和KOH处理的活性炭可储存高达7.03%的氢气[39]。Farha等人[40]所合成的MOF(NU-100)在77 K和5.6×106Pa下的储氢量则可达9.05%。更加引人注目的是,Han等[41]使用巨正则系综蒙特卡罗(GCMC)模拟对COFs进行研究(图3),发现COF-108、COF-105、COF-103和COF-102在77 K和10 MPa下分别能储氢18.9%、18.3%、11.3%和10.6%。然而,上述材料的高储氢量仅能在低温下实现; 在常温下,其储氢量往往小于1%。因此,现有物理吸附储氢材料均无法满足固体推进剂的应用要求。
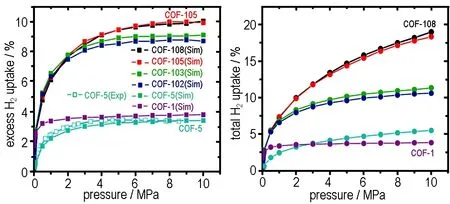
图377 K时COFs的吸氢曲线[41]
Fig.3Hydrogen adsorption curves for COFs at 77 K[41]
若要实现物理吸附储氢材料在常温乃至更高温度下的储氢,则需提高氢在材料表面吸附的焓变。研究表明,要实现常温和常压下的储氢,氢的吸附焓需达到15~25 kJ·mol-1。实现这一目标的方法有以下两种: (1)将孔径减小至亚纳米; 在亚纳米级小孔中,氢与多侧孔壁之间均存在相互作用,它们之间作用的势能场发生叠加,从而增大了吸附焓变[42]。与此相反,在孔径较大的孔中,氢只能与一侧孔壁发生相互作用,因此其对氢气的物理吸附能力较弱。研究发现,氢的吸附焓随着孔径的减小而增大,而孔径大于1.5 nm的孔对物理吸附储氢的贡献很小[43]。(2)对孔进行修饰,提高其对H2的吸附能力。对于多孔碳来说,采用某些轻元素对于进行修饰,可以增强碳基体的极性,从而提高其对H2的亲和力和吸附焓变。目前,在多孔碳中应用的掺杂元素主要有硼(B)、锂(Li)和氟(F)等。其中,B掺杂被认为是改善多孔碳吸附氢最有效的手段之一。理论计算指出,B掺杂可以将碳的吸附焓由4~8 kJ·mol-1提高至15~35 kJ·mol-1[44]; 实验研究发现,掺杂7.2%的B可以将微孔碳的吸附焓提高至12.47 kJ·mol-1[45]。对于MOFs来说,可通过使其结构中的金属离子裸露或是向其中掺杂其它金属离子两种方法进行修饰,以提高其吸附焓。Gedrich等[46]发现,通过超临界干燥处理的MOF DUT-9中有大量裸露的Ni离子,其储氢量可达4.99%,远高于Ni未裸露的类似MOF(1.1%)。此外,在MOF MIL-53中掺杂Li+离子,可以将其吸附焓由5.8 kJ·mol-1提高到11.6 kJ·mol-1,显著改善了其储氢热力学性能[47]。
与化学吸附储氢材料相比,物理吸附储氢材料的吸放氢过程不涉及活化,因而具有优异的动力学性能。然而,迄今为止,它们均只在低温下表现出较高的储氢量,究其原因在于氢吸附焓过低。现有物理吸附储氢材料的这一缺点使其在固体推进剂中的应用难以实现。然而,近年来,研究人员在提高物理吸附储氢材料的氢吸附焓方面已取得一定的进展。若能进一步进行结构和成分调节,使物理吸附储氢材料的氢吸附焓提高至能满足在室温乃至更高温度下工作的要求,物理吸附储氢材料在固体推进剂领域将具有广阔的应用前景。
5 结束语
储氢材料的放氢热力学和动力学性能显著影响其在推进剂中的应用。纳米化是一种调节材料本征性质的有效手段,在储氢材料放氢热力学和动力学的改善中获得了广泛的应用。通过机械粉碎、直接纳米合成以及纳米限域等方式可以实现化学吸附储氢材料的纳米化; 与常规材料相比,纳米化学吸附储氢材料的放氢热力学和动力学都获得了明显的改善,有利于在推进剂的燃烧过程中充分利用储氢材料中所储存的氢。对某些金属氧化物和硫化物等进行纳米化,可以改变材料与氢作用的方式,获得可提高推进剂能量并具备燃烧催化性能的化学-物理储氢材料。对物理吸附储氢材料的纳米结构和成分进行调控,则可提高其氢吸附焓,实现室温乃至更高温度下的快速吸放氢。
笔者认为,纳米储氢材料具备优异的放氢热力学和动力学性能,相比普通储氢材料在固体推进剂中有更大的实用价值。纳米储氢材料在固体推进剂中的应用的研究的重点应为: (1) 确定固体推进剂用储氢材料的放氢热力学和动力学要求,基于纳米储氢材料微观结构和形貌与其热力学和动力学性能间的关系,针对性地制备适于推进剂的纳米储氢材料; (2)与常规材料相比,纳米储氢材料普遍具有更高的活性,其与推进剂常见组分间的相容性以及对水/氧的敏感度亟需进一步研究,以满足实用化的前提要求; (3)考察纳米储氢材料对推进剂生产工艺的影响。
参考文献:
[1] 窦燕蒙, 罗运军, 李国平, 等. 储氢合金/AP/HTPB 推进剂的热分解性能[J]. 火炸药学报, 2012, 35(3): 66-70.
DOU Yan-meng, LUO Yun-jun, LI Guo-ping, et al. Thermal decomposition properties of hydrogen storage alloy/Al/HTPB propellant[J].ChineseJournalofExplosives&Propellants, 2012, 35(3): 66-70.
[2] 刘磊力, 李凤生, 支春雷, 等. 镁基储氢材料对AP/Al/HTPB复合固体推进剂性能的影响[J]. 含能材料, 2009, 17(5): 501-504.
LIU Lei-li, LI Feng-sheng, ZHI Chun-lei, et al. Effects of magnesium based hydrogen storage materials on the properties of composite solid propellant[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2009, 17(5): 501-504.
[3] Züttel A. Materials for hydrogen storage[J].MaterialsToday, 2003, 6(9): 24-33.
[4] 刘永锋, 李超, 高明霞, 等. 高容量储氢材料的研究进展[J]. 自然杂志, 2011, 33(1): 19-26.
LIU Yong-feng, LI Chao, GAO Ming-xia, et al. Progress in high-capacity hydrogen storage materials[J].ChineseJournalofNature, 2011, 33(1): 19-26.
[5] Markmaitree T, Ren R, Shaw L L. Enhancement of lithium amide to lithium imide transition via mechanical activation[J].TheJournalofPhysicalChemistryB, 2006, 110(41): 20710-20718.
[6] Liu Y, Zhong K, Luo K, et al. Size-dependent kinetic enhancement in hydrogen absorption and desorption of the Li—Mg—N—H system[J].JournaloftheAmericanChemicalSociety, 2009, 131(5): 1862-1870.
[7] Paskevicius M, Sheppard D A, Buckley C E. Thermodynamic changes in mechanochemically synthesized magnesium hydride nanoparticles[J].JournaloftheAmericanChemicalSociety, 2010, 132(14): 5077-5083.
[8] Li W, Li C, Ma H, et al. Magnesium nanowires: enhanced kinetics for hydrogen absorption and desorption[J].JournaloftheAmericanChemicalSociety, 2007, 129(21): 6710-6711.
[9] Zhu C, Hayashi H, Saita I, et al. Direct synthesis of MgH2nanofibers at different hydrogen pressures[J].InternationalJournalofHydrogenEnergy, 2009, 34(17): 7283-7290.
[10] Zhu C, Hosokai S, Matsumoto I, et al. Shape-controlled growth of MgH2/Mg Nano/microstructures via hydriding chemical vapor deposition[J].CrystalGrowth&Design, 2010, 10(12): 5123-5128.
[11] Pang Y, Liu Y, Gao M, et al. A mechanical-force-driven physical vapor deposition approach to fabricating complex hydride nanostructures[J].NatureCommunications. 2014, 5: 3519.
[12] Baldé C P, Hereijgers B P C, Bitter J H, et al. Facilitated hydrogen storage in NaAlH4supported on carbon nanofibers[J].AngewandteChemieInternationalEdition, 2006, 45(21): 3501-3503.
[13] Xie L, Zheng J, Liu Y, et al. Synthesis of Li2NH hollow nanospheres with superior hydrogen storage kinetics by plasma metal reaction[J].ChemistryofMaterials, 2007, 20(1): 282-286.
[14] Xie L, Li Y, Yang R, et al. Superior hydrogen desorption kinetics of Mg(NH2)2hollow nanospheres mixed with MgH2nanoparticles[J].AppliedPhysicsLetters, 2008, 92: 231910.
[15] Xie L, Liu Y, Li G, et al. Improving Hydrogen Sorption Kinetics of the Mg(NH2)2—LiH System by the Tuning Particle Size of the Amide[J].TheJournalofPhysicalChemistryC, 2009, 113(32): 14523-14527.
[16] Kurban Z, Lovell A, Bennington S M. et al. A solution selection model for coaxial electrospinning and its application to nanostructured hydrogen storage materials[J].TheJournalofPhysicalChemistryC, 2010, 114(49): 21201-21213.
[17] Jongh P E d., Wagemans R W, Eggenhuisen T M, et al. The preparation of carbon-supported magnesium nanoparticles using melt infiltration[J].ChemistryofMaterials, 2007, 19(24): 6052-6057.
[18] Baldé C P, Hereijgers B P C, Bitter J H, et al. Sodium alanate nanoparticles-linking size to hydrogen storage properties[J].JournaloftheAmericanChemicalSociety, 2008, 130(21): 6761-6765.
[19] Fan X, Xiao X, Shao J, et al. Size effect on hydrogen storage properties of NaAlH4confined in uniform porous carbons[J].NanoEnergy, 2013, 2(5): 995-1003.
[20] Gao J, Adelhelm P, Verkuijlen M H W. et al. Confinement of NaAlH4in nanoporous carbon: impact on H2release, reversibility, and thermodynamics[J].TheJournalofPhysicalChemistryC, 2010, 114(10): 4675-4682.
[21] Nielsen T K, Javadian P, Polanski M, et al. Nanoconfined NaAlH4; determination of distinct prolific effects from pore size, crystallite size and surface interactions[J].TheJournalofPhysicalChemistryC, 2012, 116(39): 21046-21051.
[22] Bhakta R K, Herberg J L, Jacobs B, et al. Metal-organic framework as templates for nanoscale NaAlH4[J].JournaloftheAmericanChemicalSociety, 2009, 131(37): 13198-13199.
[23] Brun N, Janot R, Sanchez C, et al. Preparation of LiBH4@carbon micro-macrocellular foams: tuning hydrogen release through varying microporosity[J].Energy&EnvironmentalScience, 2010, 3(6): 824-830.
[24] Gross A F, Vajo J J, Van Atta S L, et al. Enhanced hydrogen storage kinetics of LiBH4in nanoporous carbon scaffolds[J].TheJournalofPhysicalChemistryC, 2008, 112(14): 5651-5657.
[25] Vajo J J, Olson G L. Hydrogen storage in destabilized chemical systems[J].ScriptaMaterialia, 2007, 56(10): 829-834.
[26] Vajeeston P, Ravindran P, Fjellvåg H. Nanostructures of LiBH4: a density-functional study[J].Nanotechnology, 2009, 20: 275704.
[27] Fichtner M, Zhao-Karger Z, Hu J, et al. The kinetic properties of Mg(BH4)2infiltrated in activated carbon[J].Nanotechnology, 2009, 20: 204029.
[28] Yan Y, Au Y S, Rentsch D, et al. Reversible hydrogen storage in Mg(BH4)2/carbon nanocomposites[J].JournalofMaterialsChemistryA, 2013, 1(37): 11177-11183.
[29] Gutowska A, Li L, Shin Y, et al. Nanoscaffold mediates hydrogen release and the reactivity of ammonia borane[J].AngewandteChemieInternationalEdition, 2005, 44(23): 3578-3582.
[30] Feaver A, Sepehri S, Shamberger P, et al. Coherent carbon cryogel-ammonia borane nanocomposites for H2storage[J].TheJournalofphysicalChemistryB, 2007, 111(26): 7469-7472.
[31] Wang P, Orimo S, Matsushima T, et al. Hydrogen in mechanically prepared nanostructured h-BN: a critical comparison with that in nanostructured graphite[J].AppliedPhysicsLetters, 2002, 80: 318.
[32] Ma R, Bando Y, Zhu H, et al. Hydrogen uptake in boron nitride nanotubes at room temperature[J].JournaloftheAmericanChemicalSociety, 2002, 124(26): 7672-7673.
[33] Tang C, Bando Y, Ding X, et al. Catalyzed collapse and enhanced hydrogen storage of BN nanotubes [J].JournaloftheAmericanChemicalSociety, 2002, 124(26): 14450-14451.
[34] Oku T, Kuno M. Synthesis, argon/hydrogen storage and magnetic properties of boron nitride nanotubes and nanocapsules[J].DiamondandRelatedMaterials, 2003, 12(3-7): 840-845.
[35] Oku T, Kuno M, Narita I. Hydrogen storage in boron nitride nanomaterials studied by TG/DTA and cluster calculation[J].JournalofPhysicsandChemistryofSolids, 2004, 65(2-3): 549-552.
[36] Wan Q, Lin C L, Yu X B., et al. Room-temperature hydrogen storage characteristics of ZnO nanowires[J].AppliedPhysicsLetters, 2004, 84: 124.
[37] Pan H, Luo J, San H, et al. Hydrogen storage of ZnO and Mg doped ZnO nanowires[J].Nanotechnology, 2006, 17: 2963.
[38] Wang J, Wang L, Ma L, et al. Structures, electronic properties, and hydrogen-storage capacity of single-walled TiO2nanotubes[J].PhysicaE:Low-dimensionalSystemsandNanostructures, 2009, 41(5): 838-842.
[39] Sevilla M, Mokaya R, Fuertes A B. Ultrahigh surface area polypyrrole-based carbons with superior performance for hydrogen storage[J].Energy&EnvironmentalScience, 2011, 4(8): 2930-2936.
[40] Farha O K, Yazaydiin A O, Eryazici I, et al. De novo synthesis of a metal-organic framework material featuring ultrahigh surface area and gas storage capacities[J].NatureChemistry, 2010, 2: 944-948.
[41] Han S S, Furukawa H, Yaghi O M, et al. Covalent organic frameworks as exceptional hydrogen storage materials[J].JournaloftheAmericanChemicalSociety, 2008, 130(35): 11580-11581.
[42] Wang Q Y, Johnson J K. Molecular simulation of hydrogen adsorption in single-walled carbon nanotubes and idealized carbon slit pores[J].TheJournalofChemicalPhysics, 1999, 110: 577-586.
[43] Kowalczyk P, Tanaka H, Holyst R, et al. Storage of hydrogen at 303 K in graphite slitlike pores from grand canonical Monte Carlo simulation [J].TheJournalofPhysicalChemistryB, 2005, 109(36): 17174-17183.
[44] Kim Y H, Zhao Y F, Williamson A, et al. Nondissociative adsorption of H2molecules in light-element-doped fullerenes[J].PhysicalReviewLetters, 2006, 96: 016102.
[45] Chung T C M, Jeong Y, Chen Q, et al. Synthesis of microporous boron-substituted carbon (B/C) materials using polymeric precursors for hydrogen physisorption[J].JournaloftheAmericanChemicalSociety, 2008, 130(21): 6668-6669.
[46] Gedrich K, Senkovska I, Klein N, et al. A highly porous metal-organic framework with open nickel sites[J].AngewandteChemieInternationalEdition, 2010, 49(45): 8489-8492.
[47] Himsl D, Wallacher D, Hartmann M. Improving the hydrogen-adsorption properties of a hydroxy-modified MIL53(Al) structural analogue by lithium doping[J].AngewandteChemieInternationalEdition, 2009, 48(25): 4639-4642.
